1Hunan Institute of Traffic Engineering, Hengyang 420001, China.
2Hengyang Xin Yahan Medical Beauty Clinic, Hengyang 420001, China.
Yichun Zhang
Tel: 15616676037;
Email: 15616676037@sina.cn
Received : Nov 06, 2024 Accepted : Dec 02, 2024 Published : Dec 09, 2024 Archived : www.meddiscoveries.org
Multiple Sclerosis (MS) is an autoimmune disorder of the Central Nervous System (CNS), characterized by cytokine dysregulation, CNS inflammation, demyelination and axonal loss, which manifests mostly in young adults between ages of 20 and 30 years. The irreversible accumulation of neurological disability caused by MS imposes substantial burdens on patients, their families and society [1]. Neuroinflammation driven by CNS-resident cells, including astrocytes and microglia is pivotal in progressive MS, making these cells prime targets for therapeutic interventions [2]. Recently, it is established that neurons actively participate in initiating a Neuronal Inflammatory Stress Response (NISR) upon sensing various cytokines or stressors to maintain homeostasis. For instance, Andreadou et al. [3] revealed the neuroprotective role of IL-12 during neuroinflammation in mice is mediated by cells derived from neuroectoderm, especially neurons, rather than immune cells. Strikingly, Clark et al. [4] revealed that physiological Interferon-γ (IFN-γ) elicited a transient Signal Transducer and Activator of Transduction 1 (STAT1) activation, whereas pathological IFN-γ induced a prolonged STAT1 activation facilitated by continuous Janus Activated Kinase (JAK) activity uniquely in neurons, which can explain neurons have both homeostatic and pathological responses to IFN-γ stimulus. Besides cytokines, oxidative stress, ionic imbalances and especially glutamate excitotoxicity have been suggested as main contributing factors to neuronal cell death in MS. Excessive glutamate exposure causes excitotoxicity through intracellular calcium accumulation triggered by both activation of ionotropic glutamate receptors and calcium release from the Endoplasmic Reticulum (ER), which disturbs the balance of neuronal calcium, and ultimately leads to irreversible neuronal injury [5]. It seems cytokines and glutamate excitotoxicity are critical components in NISR, however, how these stressors interact with each other in inflammation-induced MS remains elusive.
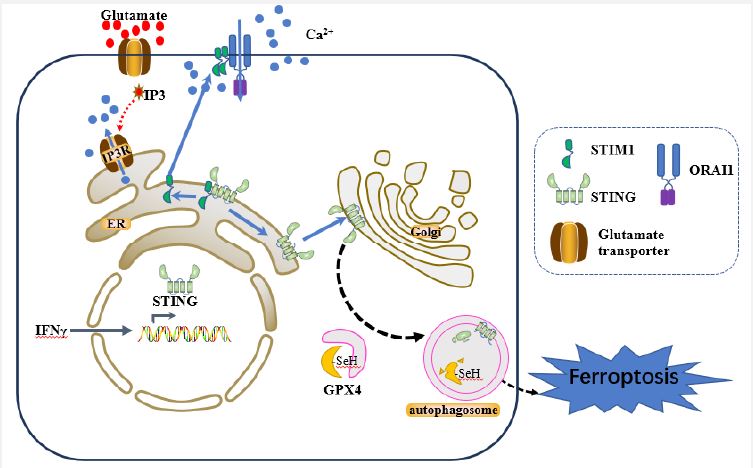
Recently, Woo et al. [6] sought to explore the underlying pathway that integrate these factors and revealed that neuronal Stimulator of Interferon Genes (STING), as a pivotal regulator, triggers autophagic-induced Glutathione Peroxidase 4 (GPX4) degradation, and subsequently initiates neurons ferroptosis, providing a target for treating inflammation-induced MS. So far, the animal model of Experimental Autoimmune Encephalomyelitis (EAE) is commonly used in investigating pathogenesis and developing new therapeutic strategies for MS due to its approximate resemblance to the key pathological features of MS [7]. Given that neuronal ER Calcium depletion (ERCa2+D) is obvious in EAE animals and in excitotoxic neuronal cell death, firstly, they discovered that neuronal Stromal Interaction Molecule 1 (STIM1, an ER calcium sensor protein) deficiency mice exacerbated neuronal damage, indicating STIM1 is necessary for neuronal resilience during EAE neuroinflammation [6]. Furthermore, STIM1 translocated from neuronal somata to injured axons, representing a neuronal excitotoxic damage in EAE mice. Subsequently, they found glutamate excitotoxicity induced ERCa2+D and prompts STIM1 to translocate from ER to plasma membrane, interacting with ORAI1 (a Calcium Release-Activated Calcium (CRAC) channel), which allows Ca2+ entry from the extracellular space and increases cytosolic Ca2+ levels in neurons [6]. In line with this findings, Witte et al. [8] also found cytoplasmic Ca2+ accumulations drives axon degeneration in a MS model with calcium imaging technique. Intriguingly, STIM1 deficient neurons showed no differences with Wild Type (WT) neurons in susceptibility to glutamate excitotoxity. However, they are more susceptible to glutamate compared with WT neurons under IFN-γ exposure [6], hypothesizing an inflammation-induced factor is required for the increased vulnerability shown in Stim1-conditional Knockout (c-KO) EAE mice.
STING is an ER signaling adaptor that has been recognized in the immune response against pathogens activated by the cyclic GMP-AMP synthase (cGAS) after detecting foreign double-strand DNA (dsDNA). In addition to its involvement in host defense, aberrant activation of STING is linked to the pathology of autoimmune diseases, inflammatory diseases and neurodegenerative diseases [9]. Strikingly, Woo et al. [6] discovered Sting1 is the most strongly induced neuronal gene both in in vitro (neurons exposure to IFN-γ) and in vivo (EAE mice) models, highlighting that STING is specifically expressed in inflamed neurons. Furthermore, co-immunoprecipitation assay showed STIM1 directly interacts with STING in the neuronal ER membrane, maintaining STING inactive under steady state, which is consistent with the result in the previous study that STIM1 is an ER anchoring factor to suppress the activity of STING [10]. However, treatment with DMXAA (a STING ligand) or ERCa2+D, as well as genetic deletion of Stim1, each stimulus result in STING dissociating from STIM1. This dissociation causes STIM1 trafficking to cell membrane, while STING is activated to its phosphorated form (p-STING) and translocate to the Golgi compartment [6]. Moreover, neurons overexpressing STING or neurons exposed to prolonged IFN-γ were more vulnerable to glutamate excitotoxicity, which was rescued by treating STING inhibitors C176 or H151 [6]. This observation confirmed the hypothesis mentioned above, namely, STING is the inflammation-induced factor responsible for the STIM1-dependent neuronal vulnerability.
Unexpectedly, this STING activation in neurons initiates a non-canonical STING signaling, which in turn leads to autophagy and amplify neuronal injury [6]. Accumulating evidence suggested that autophagy is involved in regulating iron content, ROS production, and lipid peroxidation during ferroptosis [11]. In this project, neurons overexpressing STING or neurons subjected to IFN-γ markedly increased autophagy-driven degradation of GPX4, which was reversed by treatment with STING inhibitor H151 or autophagy inhibitors SAR and SBI. Consistently, with the decreased GPX4 concentration, STING-mediated autophagy increased Reactive Oxygen Species (ROS) levels and thereby promoting ferroptosis after glutamate challenge [6] (Figure 1).
Growing evidence substantiated ferroptosis is a contributor to glutamate excitotoxicity, neuronal dysfunction, and MS disease progression [12,13]. As reported by Rothammer et al. [12], G9a, a histone methyltransferase reduced anti-ferroptotic genes expression and triggered ferroptosis in MS brain tissue, while G9a inhibitor UNC0642 increased anti-ferroptotic genes expression, reduced neuronal loss and in turn improved clinical outcome. Similarly, San et al. [13] demonstrated that administering the ferroptosis inhibitor UAMC-3203 significantly delays relapse and alleviates disease progression in a relapsing-remitting MS model, highlighting ferroptosis as a detrimental factor in MS pathology. Here, the neuronal STING-GPX4-ferroptosis pathway provides many intervention targets to halt inflammation-induced neurodegeneration. As anticipated, both genetic (Sting1-cKO mice) and pharmacological interventions with STING antagonists (C176 or H151) decreased autophagy, increased GPX4 levels, reduced ferroptosis in neurons, and accordingly, protected against inflammation-induced neurodegeneration. As a result, it altered the neuronal environment’s response, illustrated by an increase in homeostatic microglia with high expression of Triggering Receptor Expressed on Myeloid cells 2 (TREM2) following the neuron-specific Sting1 deletion [6].
Notably, the activation of canonical STING signaling in microglial plays a crucial role in the progress of various neurodegenerative diseases, while STING inhibitors offer multiple benefits in promoting a healthy CNS and significantly alleviates inflammation and neurodegeneration [14]. Moreover, STING was found to be upregulated in brains of individuals with MS [12,6], therefore, targeting STING holds great promise for management of this neurodegenerative disorder.
In summary, this project summarizes IFN-γ and glutamate jointly induce the NISR in MS, highlighting STING is a vital regulator of the detrimental NISR and is a potential therapeutic target. However, there are few points that should be taken into consideration. First of all, further investigation on the association between STING activation and pathological autophagic-induced GPX4 degradation may help to identify novel therapeutic targets. Second, glutamate excitotoxicity alongside chronic IFN-γ exposure was employed to construct in vitro model of MS, which notably simplified the complexity of inflammation-induced MS. Hence, more convincing in vivo models reflecting inflammation-induced demyelination and neuronal loss are required in future research. Last but not least, it will be beneficial to determine the optimal dosage of STING inhibitors, since complete blocking STING signaling in human would cause adverse effects by increasing susceptibility to infection. Collectively, a better comprehension of the potential mechanism could facilitate the utilization of STING-based treatments in individuals with MS.
Acknowledgments: This work was supported by the Hunan Provincial Natural Science Foundation of China (No: 2019JJ40077).