1Faculty
Ibtissam El Oualic
Email: btissam.elouali94@gmail.com
Received : Oct 09, 2024 Accepted : Nov 06, 2024 Published : Nov 13, 2024 Archived : www.meddiscoveries.org
Biliary atresia is a rare disease of unknown etiology characterized by an obstruction of biliary ducts occurring during the prenatal period due to progressive fibrosis of extrahepatic biliary ducts. Surgery is the only way to prevent hepatic cirrhosis which unique treatment is a liver transplant. It can be associated with other abnormalities grouped into a syndrome called biliary atresia polysplenic malformation syndrome which changes the surgical anatomy making the surgery very challenging. Imaging especially ultrasonography is key in the preoperative assessment of patients suspected of biliary atresia. We are describing the case of biliary atresia poly splenic malformation syndrome with polysplenia, dorsal pancreatic agenesis, preduodenal portal vein, and reverse rotation of the intestine in a 26-year-old boy who underwent Kasai surgery for cystic biliary atresia on day 6 of life, who presented years later for intermittent cholangitis on biliary cirrhosis, depicting the importance of imaging in making early diagnosis and follow up.
Keywords: Biliary atresia; Polysplenia; Kasai procedure; Jaundice.
The biliary atresia splenic malformation syndrome encompasses a group of extra hepatic anatomical abnormalities essentially polysplenia, intestinal malrotation or abdominal situs inversus and congenital cardio-vascular and pulmonary malformation in association with extra hepatic biliary atresia. Other associations have been less frequently reported in history, such as renal, pancreatic and choledochal anomalies or congenital trachea-esophageal fistula [1]. The mechanism behind this malformation syndrome is thought to be genetic defect in the first trimester of embryogenesis leading to aberrant development of organs either defective or excessive organogenesis [2]. The radiological preoperative workup in biliary atresia must be exhaustive to detect these anomalies, which can be detrimental for the patient if not mentioned to the surgeon [3].
We report the case of a 26-year-old boy who underwent Kasai surgery for cystic biliary atresia on day 6 of life with the creation of an end-to-side hepato-porto-enterostomy to form the Roux-en-Y structure.
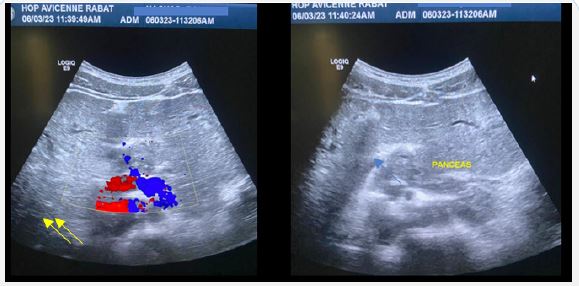
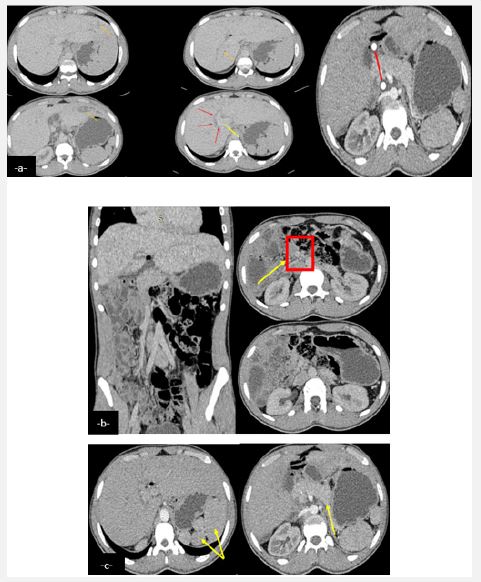
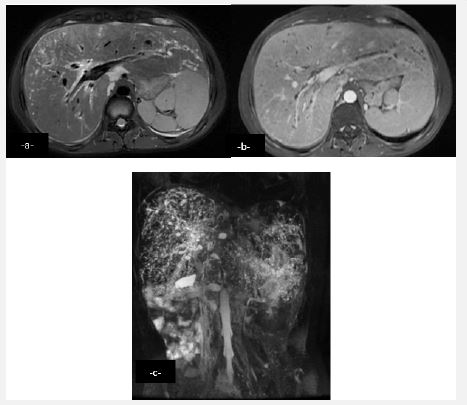
Years later the patient presented with intermittent episodes of jaundice without fever with dark urine and normal colored stools. Clinical examination noted hepato-splenomegaly. Hepatic workup showed rise in liver enzymes (Alanine Transaminase (ALT) = 250 U/L and Aspartate Aminotransferase (AST) = 100 U/L) with cholestasis (Total bilirubin = 70 mmol/l and direct bilirubin = 50 mmol/l). An ultrasound found a dilation of the intrahepatic bile ducts more marked in the periphery, a dilated preduodenal portal vein, associated with a malformation syndrome encompassingpoly splenia, dorsal pancreatic agenesis and a transposition of mesenteric vessels suggestive of complete intestinal malrotation (Figure 1). Abdominal scan demonstrated the same findings with a complete malrotation of intestine (Figure 2). MRI allowed a better analysis of the bilio-digestive anastomosis which was free of any thickening. Moreover, it showed signs of biliary cirrhosis with peribiliary cysts associated with biliary atresia non cardiac polysplenic syndrome (Figure 3). This sub icteric episode, eventually related to either mild cholangitis or transient stenosis of the biliodigestive anastomosis, resolved completely after a week. The patient remained asymptomatic on two-weeks follow up.
Biliary Atresia (BA) is a rare life-threatening inflammatory fibro-obliterative cholangiopathy affecting intra and extrahepatic bile ducts in neonates and infants advancing, if not treated early, to end-stage liver disease, with an incidence of approximately 1/8000-1/18000 [4]. It is unusually associated with laterality malformations known as embryonal or syndromic biliary atresia in 10-25% of cases [5], and the most common association is the Polysplenia syndrome which is reported in about 10% of cases and can include situs inversus inferior vena cava, and cardiac anomalies. This special subgroup of biliary atresia is reported to have a poorer prognosis.
On one hand, the exact etiopathogenesis of biliary atresia itself has not been fully elucidated and it might be consequent to sclerosing inflammation of unknown etiology. On the other hand, the polysplenic malformation syndrome might be involved in an entirely different process occurring at around fifth week of gestation when organogenesis happens [6], and this is supported by studies that showed that the probability of this association is higher in neonates with early-onset biliary atresia [7]. Several factors have been implicated in the pathogenesis of biliary atresia syndrome; Genetic factors contributes by and large in the pathogenesis. Mutations in a small number of ciliopathyand laterality genes, including Cryptic Family 1 (CFC1) andpolycystic kidney disease 1 like 1 (PKD1L1) has been frequently reported [8,9]. In addition to that, abnormal intrauterine environment, such as maternal diabetes has also been incriminated in some cases [10]. The diagnosis of polysplenia syndrome is often made during first months of life. Its association with biliary atresia requires early diagnosis and treatment to prevent progression to cirrhosis [11]. Prenatal diagnosis is possible thanks to the contribution of obstetrical ultrasound, by visualizing certain elements of the polysplenia syndrome, especially subhepatic cysts that are highly suggestive of either cystic biliary atresia, the surgical neonatal emergency, or a choledochal cyst [12].
After birth, polysplenia malformation syndrome manifest with cholestatic jaundice with elevated conjugated bilirubin in biological screening [13] and bowl obstruction due to digestive atresia. Fortious diagnosis of BASM during congenital heart disease assessment is also possible [14]. Therefore, the preoperative workup should not be limited to hepatobiliary screening in order to detect these abnormalities which impact the initial portoenterostomy and may impede eventual orthotopic liver transplantation that might follow this surgery.
US is the key tool in the preoperative workup. Careful abdominal sonographic assessment reveals echogenic fibrous tissue anterior to the portal vein representing remnant extrahepatic bile duct which we refer to as the triangular cord sign [15], dilated hepatic artery, gallbladder ghost triad; atretic gallbladder with lobular contour and indistinct wall. It can depict signs of liver cirrhosis and an eventual choledochal cyst. Other vascular anomalies associated with syndromic biliary atresia can be found like polysplenia, preduodenal portal vein positioned anteriorly to the superior mesenteric artery and aorta. Longitudinal sonogram shows absence of intra hepatic portion of inferior vena cava and hepatic veins enter the right atrium directly indicating an interrupted inferior vena cava [16].
Ultrasound (US) signs have a high specificity, although a normal US examination does not rule out biliary atresia. Hepatobiliary scintigraphy using radiotracers demonstrates in patients with biliary atresia a normal hepatic uptake without evidence of excretion into the bowel at 24 hours [17]. Liver biopsy may be helpful in determining whether surgery is necessary If biliary atresia diagnosis is very likely, Cholangiography (percutaneous or retrograde endoscopic) is scheduled and contributes in positive diagnosis by demonstrating the absence of biliary ducts. If there is no flow of contrast into the extrahepatic biliary ducts, a diagnosis of biliary atresia is made and a Kasai procedure is performed at that time. CT-scan is not indicated in initial management of biliary atresia. However, post operatively, it helps assess the porto-entrostomy and depict surgical complications such as bile lakes, which is the most common finding, associated with worse prognosis including increased incidence of cholangitis. requiring prophylactic antibiotics, and sometimes, percutaneous bile drainage or surgical refashioning management [18]. MRI’s major role is evaluating the liver parenchyma in later follow up. It has a significant role in screening cirrhotic livers for small hepatocellular carcinomas, assessment and classification of cystic dilatation of the intra hepatic biliary system (type A, noncommunicating cyst; type B, cyst with tiny communication with the intestinal loop; and type C, cystic dilatation) frequently described around 4 to 10 years after the Kasai operation [19,20]. Informing preoperatively the surgeon of these abdominal abnormalities is crucial in order to render surgery easier and more efficient [21]. Unnotified preduodenal portal vein may lead to itsinadvertent ligation [22,23]. Also, a gastrointestinal malrotation, complicates the construction of the conduit [23]. Furthermore, a preduodenal portal vein, an interrupted inferior vena cava, and an anomalous hepatic artery constitute abnormal connections to the native liver. Subsequently, hepatic transplantation is bound to fail resulting in the deaths of the children as reported in many series [22].
Biliary atresia polysplenic malformation syndrome is seen in a significant number of infants with biliary atresia.
An exhaustive sonographic assessment is recommended in the preoperative workup and for follow-up of these patients in order to depict hepatic complications, especially biliary cirrhosis.
Acknowledgement: I would like to express my gratitude to my professors and all the colleagues who participated in the completion of this work. The authors declare that they have no competing interests.
Conflicts of interests: The authors declare that they have no conflicts of interest.
Ethics statement: Clinical presentations are anonymized and there are no images to identify patients.
Written consent of the patient was obtained for this study.
Author’s contributions
Ibtissam El Ouali: Conception of the work, design of the work and acquisition of data.
Kawtar Imrani: Acquisition of data and revising the work.
Kenza Berrada: Acquisition of data.
Hiba Zahi: Acquisition of data.
Nabil Moatassim Billah: Revising the work critically for important intellectual content.
Ittimade Nassar: Revising the work critically for important intellectual contentand final approval of the version to be published.
Funding: There was no source of funding.