Department of Medical Biology, University of Québec at Trois-Rivières, Trois-Rivières, QC G8Z 4M3, Canada.
Patrick Narbonne
Tel: +1-819-376-5011;
Email: patrick.narbonne@uqtr.ca
Received : Nov 29, 2023 Accepted : Dec 15, 2023 Published : Dec 22, 2023 Archived : www.meddiscoveries.org
The p38 Mitogen Activated Protein Kinase (MAPK) is a key member of a highly conserved superfamily of protein kinases. Mostly sensitive to inflammatory cytokines and stress signals, p38 activation leads to a variety of cellular responses, including proinflammatory cytokines secretion, cell survival, apoptosis induction and the regulation of cell proliferation. Here, we carried a focused investigation on the recent literature, across all systems and organisms, concerning the roles of p38 in the regulation of Stem Cell (SC) proliferation. Interestingly, we found that p38 can exert either a pro- or an anti- proliferative function, depending on the specific models and contexts. It is also activated by a variety of upstream signals and regulates SC proliferation through a wide diversity of molecular effectors. Whereas p38 regulates SC proliferation mostly cell-autonomously, an example of a non-cell autonomous p38-dependent regulation of SC proliferation was recently uncovered. Altogether, bringing together these recent findings improve our comprehension of the effects p38 has on SC proliferation, while these mechanisms and effectors appear as being conserved through evolution.
Keywords: Pneumonia; Sepsis; Shock; Intensive Care Unit-Acquired Weakness; Myasthenia; Complications; Prognosis.
Mitogen Activated Protein Kinases (MAPKs) are a highly conserved superfamily of Ser/Thr kinases involved in signal transduction. The MAPK signaling cascade requires consecutive phosphorylation of three protein kinases: MAPK kinase kinase (MAP3K) phosphorylates MAPK kinase (MAP2K), which in turn phosphorylates MAPK. Following extra and/or intracellular signals, the activation of MAPKs induce a wide range of cellular responses by phosphorylating many downstream effectors. The p38 MAPK (p38) is one of the main MAPK families. The canonical p38 pathway mainly responds to inflammatory cytokines and stress signals, leading to the activating phosphorylation of one of several MAP3Ks (MEKK1-3, MLK2/3, ASK, Tpl2, TAK1 and/or TAO1/2), in turn activating a MAP2K (MKK3/6), to finally turn ON the p38 MAPK. Following this activation, p38 phosphorylates a number of downstream effectors, including cPLA2, MNK1/2, MK2/3, HuR, GADD153, Ets1, p53 and MSK1/2 in the cytoplasm, and ATF1/2/6, MEF2, Elk-1, GADD153, Ets1, p53 and MSK1/2 in the nucleus. The activation of p38 most often results in the production of proinflammatory cytokines, the promotion of cell survival or apoptosis, and the regulation of cell proliferation [1].
In this minireview, we aimed to cover the effects of p38 signaling on Stem Cell (SC) proliferation, across all systems and organisms. We conducted a PubMed search using the terms “p38 stem cell proliferation” and focused on articles published since 2020. Among these, we selected the articles that we considered as being the most relevant and informative regarding this important question.
Several pieces report a pro-proliferative role for p38 in various types of SCs. We first present In vitro evidence, largely relying on SC proliferation assays in culture, followed by in vivo analyses that quantified SC proliferation within complete organisms.
In vitro analyses
Several recent In vitro analyses have placed p38 as an important positive regulator of SC proliferation. In cultured murine vascular SCs for example, the addition of ATP activates purinergic metabotropic P2Y receptors and promotes p38 activation. This was accompanied by an increase in the number of viable cells compared to an unstimulated control. Interestingly, the addition of the p38 inhibitor SB203580 decreased both phosphorylated p38 (p-p38) levels and viable cell numbers, suggesting that p38 promotes vascular SC proliferation in response to ATP [2].
Another example of the pro-proliferative functions of p38 In vitro comes from an analysis of chicken muscle SCs. In chicken muscle SCs, gga-miR-3525 microRNA has an anti-proliferative function: its overexpression inhibited SC proliferation, whereas its depletion promoted SC proliferation. Indeed, the inhibition of miR-3525 decreased the proportion of cells in G0/ G1 phase, and increased the proportion of cells in S phase, suggesting an increased proliferation of muscle SCs. PDLIM3, an inhibitory target of miR-3525, had the opposite effect, and promoted SC proliferation. Notably, PDLIM3 overexpression in chicken muscle SCs increased p38 activation. This activation was abrogated either by mir-3525 overexpression, or by exposure to the p38 inhibitor SB203580, suggesting PDLIM3 positively regulates p38, and mir-3525 inhibits p38 by targeting PDLIM3 [3]. Furthermore, in murine cultured myoblasts, MicroRNA-668-3p treatment directly downregulates its target, Appl1, a positive regulator of myoblast proliferation and differentiation. Noticeably, miR-668-3p inhibition and Appl1 overexpression are accompanied by an increase in p-p38 levels in the same cell type, suggesting these actors play in the same cascade to promote myoblast, and potentially also muscle SC proliferation [4]. These two studies emphasized disruption of p38 pro- proliferative functions by microRNA-dependent inhibition of its upstream effectors.
The p38 MAPK also regulates SC proliferation downstream extracellular stress signals. Pulsating fluid shear stress is a model where culture medium is pumped through a flow chamber containing SCs, submitting them to a cyclic changing pressure gradient. In cultured murine primary muscle SCs, also known as satellite cells, pulsating fluid shear stress promotes p38 phosphorylation and is associated with the upregulation of c-Fos, Cdk4 and IL-6 proliferation marker mRNA levels [5]. As previous studies had demonstrated that p-p38 was required for muscle SC proliferation [6], these results raised the hypothesis that pulsating fluid shear stress could promote muscle SC proliferation through p38 activation.
The p38 MAPK promotes proliferation in other mammalian SC culture models. The treatment of rabbit corneal epithelial SCs, also known as limbal SCs, with pro-proliferative glycoprotein SPARC quickly induced p38 phosphorylation. Enhanced SPARC-mediated proliferation was abolished following a cotreatment with the p38 inhibitor SB203580, or the JNK MAPK inhibitor SP600125, indicating that p38 and JNK MAPK pathways are positive regulators of SPARC-mediated limbal SC proliferation [7].
The positive regulation of SC proliferation by p38 is conserved across several cultured human SC types. PBK/TOPK is a MAP2K-like protein that is activated during M- phase in HeLa cancer SCs by cyclinB/Cyclin Dependent Kinase 1 (CDK1) phosphorylation [8]. While PBK/TOPK has been shown to phosphorylate P38 In vitro, but not ERK or JNK MAPK, elevated p-p38 levels were detected in cyclinB+ primary cerebral granule cell precursor, suggesting that p38 is activated during mitosis in these progenitors too. Furthermore, the p-PBK/ TOPK and p-p38 signal overlapped in primary cerebral granulecell precursor lines. Treating these cerebral granule cell precursors with the p38 inhibitor SB203580 partially decreased BrdU+ S-phase cells, again supporting the pro-proliferative function of this pathway in neural progenitors [9].
Treating human cardiac SCs with blood serum and blood plasma increased cell counts, suggesting increased proliferation rates. Treatment with either of two p38 inhibitors, BMS-582949 or SB20239063, partially disrupted the pro-proliferative and pro- survival effect of blood serum exposure [10].
Still in humans, the co-inhibition of p38 and mTORC1 with inhibitors LY2228820 and rapamycin, respectively, downregulated cultured umbilical cord blood SC proliferation, and increased G0/G1 phase cell counts [11]
More recently, in human neural SCs, as well as in derived cerebral organoid cells, it was found that the SKF83556 ligand activates the dopamine D1 receptor and its two intracellular effectors β-arrestin 2 and Epac2. β-arrestin 2 in turn activates p38, which cooperates with Epac2 to promote proliferation [12].
Finally, the sympathetic nervous system activates the ADRA1B adrenergic receptor at the surface of dental pulp SCs and triggers their metabolic reprogramming from oxidative phosphorylation to anaerobic glycolysis metabolism, all while inhibiting SC proliferation. Interestingly, the levels of p-P38 were found to be reduced following ADRA1B overexpression and increased following ADRA1B loss-of-function. Moreover, dental pulp SC treatment with a p38 agonist, dehydrocorydaline chloride, was sufficient to rescue metabolic reprogramming and the weakened proliferative capacity that result from ADRA1B overexpression. Altogether, those results suggest that ADRA1B inhibits p38 to reduce dental pulp SC proliferation [13].
In vivo analyses
In vivo examples of positive regulation of SC proliferation by p38 were found in an invertebrate organism. The parasitic flatworm Schistosoma mansoni adults release mediators promoting their survival while they infect a human host, as they are surrounded by the host’s immune cells and effectors [14]. These excretory-secretory products that are released by both male and female individuals activate p38 signaling in the tegument, musculature, and reproductive organs of opposite sex individuals, and promote germline development. As a matter of fact, single-sex cultures displayed decreased germline SC proliferation while exposure to excretory-secretory products restored high germline SC proliferation rates. Treatment with the p38 SB203580 inhibitor completely abolished the increased proliferation, indicating that this mechanism depends on p38 activation. These results are consistent with the robust p38 expression in S. mansoni germline SCs and their cellular progeny that was reported more recently [15].
The p38-dependent stimulation of SC proliferation downstream of stress signals is conserved in mammals. Under hematologically stressful conditions, such as during irradiation, the body becomes depleted of its blood cells. If a lethal dose is not reached, bone marrow hematopoietic SCs enter the cell cycle, and repopulate the entire hematopoietic system [16]. Similarly, following 5-fluorouracil treatment, differentiated hematopoietic cells are systemically eliminated and proliferation of quiescent hematopoietic SCs is induced. In such conditions, the mortality of p38 (-/-) mice was significantly higher than that of control mice, suggesting that p38 function is required to promote hematopoiesis, required to ensure survival. Accordingly, p-p38 expression was significantly increased in hematopoietic SCs following 5-fluorouracil treatment. The p38 (-/-) hematopoietic SCs transplanted in lethally irradiated animals displayed lower proliferation rates compared to animals transplanted with wild-type cells. In the same study, authors also studied the mechanisms by which transplanted p38 (+/+) hematopoietic SCs replace damaged endogenous hemotopoietic SCs in lethally irradiated mice. They found that in these cells, after transplantation, p-p38 activates the Mitf transcription factor, leading to increased Impdh2 expression. The IMPDH2 protein in turn was found to positively regulate purine metabolism, promoting cell cycle progression [17].
Murine spermatogonial SC treatment with FGF9 increases p38 phosphorylation and may also promote proliferation, since it leads to increased cell numbers relative to untreated cultures. The inhibition of p38 with SB203580 reversed the phenotype induced by FGF9, suggesting that p38 mediates the proproliferative effects of FGF9 in this system [18]. Interestingly, authors found that Fgf9 overexpression inhibits spermatogenesis in mice and leads to undifferentiated spermatogonia accumulation. Therefore, FGF9 may not only promote spermatogonial SC proliferation, but also inhibit their differentiation.
Still in mice, a Casp9 floxed allele was generated and crossed into a line driving inducible Cre recombinase expression in the hair follicle SC compartment (bulge and hair germ). These transgenic mice also displayed a Rosa26-EYFP cassette, allowing the tracking of Cre recombination events. During the telogen phase of hair growth, Casp9 depletion in hair follicle delayed apoptosis and the clearance of hair follicle SCs. Casp9 depletion also significantly increased both EYFP+ and EYFP- hair follicle SC proliferation in bulge and hair germ compartments. The majority of proliferating Ki67+ cells were however adjacent to cleaved-Casp3+ apoptotic cells, the latter also expressing the Wnt3 pro-proliferative ligand. This Wnt3 mRNA induction in apoptotic cells was abolished by a treatment with the p38 inhibitor SB203580. Interestingly, the Dusp8 dual-specificity phosphatase, a physiological inhibitor of p38, can be cleaved by Casp3 In vitro. Moreover, Dusp8 protein levels were reduced in cleaved-Casp3+ hair follicle SCs. This low level of Dusp8 corelated with high Wnt3 expression and p38 phosphorylation. Mechanistically, it was therefore proposed that Casp3 may cleave the Dusp8 phosphatase in apoptotic cells to alleviate the inhibition of p38, thus promoting Wnt3 induction and the stimulation of adjacent hair follicle SC proliferation. This study shows for the first time, a non-cell autonomous pro-proliferative function of p38, through Wnt3 ligand induction [5,19].
Again, we will first cover the In vitro situations where p38 had anti-proliferative effects, followed by in vivo evidence.
In vitro analyses
In organoids derived from murine intestinal crypt cells, hexokinase 2 depletion, or 2-desoxyglucose treatment, inhibits glycolysis. As a result, organoids growth was significantly decreased compared to untreated controls, and this phenotype was rescued by adding the SB202190 p38 inhibitor. Moreover, in hexokinase 2-depleted and 2-desoxyglucose-treated organoids, p38 inhibition led to the downregulation of differentiation marker expression, such as LYZ, MUC2 and ATOH1. These results suggest that under metabolic stress condition, p38 inhibits intestinal crypt SC proliferation, instead promoting their differentiation [20].
Negative regulation of SC proliferation by p38 also occurred in rodent cultures. During cardiac muscle differentiation in culture, their proliferation stalls. Exposure to the p38 inhibitor SB203580 however abolished this cell cycle arrest by removing Raf/ERK inhibition and led to increased proliferation in a pro-differentiation culture medium. Hence, it was proposed that p38 promotes cell cycle arrest by inhibiting the Raf/ERK pathway [21]. This result is further supported by a recent study of human embryonic SCs. In these, p38 inhibition by SB203580 led to increased cell counts compared to control, likely due to increased proliferation [22].
The sometimes anti-proliferative role of p38 was confirmed in another model of cultured mammalian muscle SCs. Short term p38 inhibition by SB203580 decreased proliferation in cultured bovine muscle SCs. The p38 inhibitor also caused an increase in muscle SC marker PAX7 mRNA expression and decreased muscular cell differentiation markers pHSP27 and MyHC at the protein level. However, at lower inhibitor concentrations and in the presence of FGF-2, p38 inhibition slightly increased cell numbers compared to non-treated cells. These results suggest that p38 signaling could have both pro- and anti-proliferative functions in the same cell type and species depending on the context, on top of influencing cell identity [23].
In vivo analyses
In vivo evidence showing p38-dependent inhibition of SC proliferation were hardly found across the reviewed literature: this is clearly an area where further investigation will be required.
Yet, in the mouse intestine, aging was associated with increased MKK6 MAPKK expression, increased p38 activation, and increased mTORC1 activity. Rapamycin treatment suppressed both MKK6 and p38 activation, suggesting that mTORC1 acts upstream of the MKK6 to p38 axis. In addition, p38 inhibition with SB203580 increased Ki67+ intestine transit amplifying cells, supporting an anti-proliferative effect of p38 in this in vivo system [24]. Whether intestinal SCs are similarly affected by p38 was however not investigated.
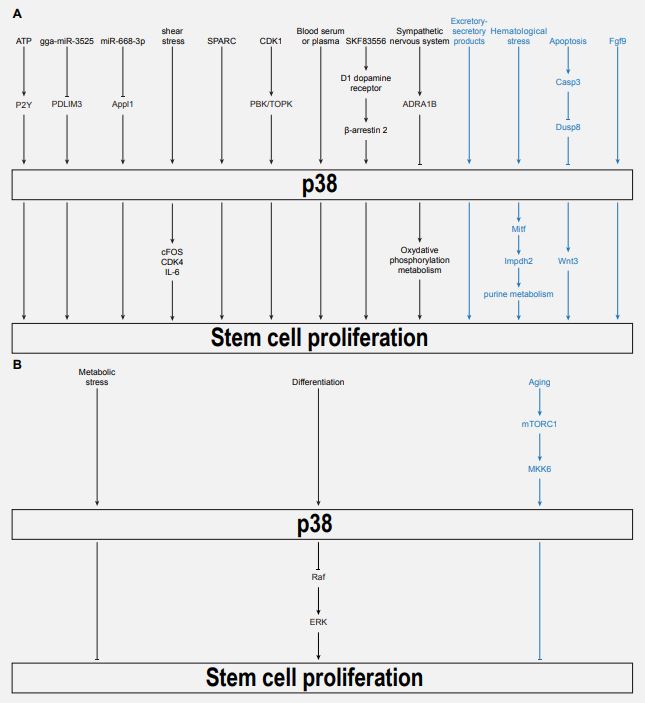
The effects of p38 on SC proliferation were investigated in recent studies across a wide variety of models and organisms. These are summarized in Figure 1. It appears that the particular effect, whether it be promoting or inhibiting SC proliferation, is highly dependent on the model and context. In most cases, the effects were cell-autonomous, but there was also an example where p38 promoted SC proliferation non-autonomously, from adjacent cells. Upstream regulators and downstream effectors also differed depending on the cell types and contexts. In many cases, it was unclear whether p38 regulation required canonical upstream regulators (MAP3K and MAP2K) and whether it was acting through the same downstream targets. It also appears that p38 can even have opposite roles in the same cell types, depending on species and contexts (e.g., muscle SCs; [3-5,23], and even within the same cells, depending on its activation level [23]. Addressing the full molecular cascades leading to p38-mediated inhibition of SC proliferation within their in vivo complexity is certainly one of the next challenges in the field.
Acknowledgements: Research in the Narbonne Laboratory is funded by grants from the NSERC (RGPIN-2019-06863, RGPAS-2019-00017, DGECR-2019-00326) and the CIHR (PJT169138) to PN.
PN is an FRQ-S Junior 2 Bursary Scholar (310643).