1Zhenjiang 4th People’s Hospital Affiliated to Jiangsu University, Jiangsu 212000, China.
2Medical School, Jiangsu University, Jiangsu 212000, China.
Rui Ma
Email: jsnjmr@163.com
Received : Sep 08, 2023 Accepted : Oct 03, 2023 Published : Oct 10, 2023 Archived : www.meddiscoveries.org
Objective: To accumulate clinical features and clinical laboratory data about cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy.
Materials: In this study, 2 cases and their families with cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy admitted to our hospital were engaged for retrospective study.
Results: CADASIL is a familial disease, and different members of the same family had different manifestations. Diseases can be caused by mutations at different loci. Migraine, subcortical cerebral infarction and dementia appear regularly at different phases of ages. They are typical clinical manifestations of CADASIL. Subcortical cerebral infarction, especially in the external capsule area and corpus callosum in middle age, is more characteristic.
Conclusion: There is an autosomal dominant inheritance principle in CADASIL family. Gene diagnosis is the primary and preferred diagnostic basis for CADASIL. The mutation sites related to CADASIL need to be explored. The relationship between mutation and pathological changes and more studies are needed to understand the characteristics of the disease.
Keywords: Cerebral autosomal dominant arteriopathy with subcortical infarcts; Leucoencephalopathy; Diagnosis; Infract.
The natural history of Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leucoencephalopathy (CADASIL) has obvious rules in different stages [1,2]. Although CADASIL patients tend to develop the disease at an early age, symptoms gradually develop until about age 50, followed by infarct dementia and psychiatric symptoms in the elderly. Similar to other rare diseases, which are misdiagnosed and the incidence is underestimated because of limited clinical data, knowledge and diagnostic techniques. Increased data collection on the disease is important for improving our understanding of the disease and diagnosis and treatment [2-4].
Case 1 is a 48-year-old married woman with “two eyes for 2.5 months and 20 days of unclear teeth. On December 10, 2015, he entered the neurological department of Jiangsu University’s Fourth People’s Hospital. The patient was admitted to our hospital in April 2014 due to numbness of the left apex and upper extremities and weakness of the extremities. Two and a half months ago, the patient presented with sudden comatose with bilateral vision and dyskinesia of both eyes. She was hospitalized in the neurologic department and diagnosed as having multiple sclerosis. He was speechless for 20 days, coughed when drinking water, had numbness to his fingers and back, and walked unstablely.
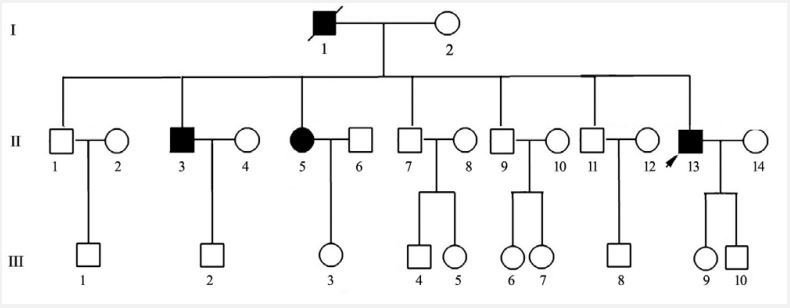
Family history: See Figure 1. His father had a history of migraine and cerebral infarction, which began at age 65 with recurrent cerebral infarction, leaving hemiparesis, long in bed, and died at age 72. The patients had five brothers and one sister, one of whom had a history of migraine.
Case 2, a 66-year-old married man who had had dizziness and vomiting for 7 years and 10 hours. Graduated on October 30, 2018. The patient in 2011 had dizziness attack without obvious predisposing factor, which manifested as dizziness with apparent nausea and vomiting, and could be improved by lying down for half a day without opening the eyes. In 2011, approximately 1 attack occurred in a half-month period, with each symptom slightly identical, for a total of 10 attacks; there have been no apparent attacks in the years since. These dizziness attacks began again this year, and in May I came to the outpatient clinic to receive flunarizine capsules orally, without an attack for several months. She reappeared on October 10 and improved after a rest. She woke up this morning with dizziness, nausea and vomiting, and did not improve after rest.
Previous history: Right-ear hearing loss from childhood; History of appendicitis and thyroid cyst surgery; History of migraine at a young age with prominent visceral neurological symptoms of nausea and vomiting at onset; History of cerebral infarction for 6 years, when the right extremity was weak, with no sequelae after treatment. There was one episode of dizziness that was diagnosed by auscultation as atrial fibrillation in a local clinic without ECG confirmation.
Family history: The patient’s parents died of cerebral infarction for more than 10 years, and his father was demented; the patient had two sisters and one sister, of whom the older sister had a history of cerebral infarction, dementia, and the younger sister had a history of cerebral infarction.
Case 1 underwent heat sequencing of the NOTCH3 mutation in CADASIL disease (Nanjing Jinyu Medical Examination Institute): a heterozygous causative mutation was detected: NOTCH3 cDNA level: c. 421C>T protein level: p. (Arg141Cys), diagnosis: CADASIL (Figure 2).
Examination: Diplopia, small fissure in the right eye, inability of the right eye to adduct when both eyes gaze to the ipsilateral level, abduction of the left eye, rotational nystagmus in the left eye, incomplete limited upward and downward movements in the right eye, limited upward movements in the left eye, nystagmus at the level of the right eye; shallow left lip groove, extension of tongue to the left, dysarthria, dull pharyngeal reflexes, bilateral tendon reflexes, right ankle cramps, none of the bilateral Babinski’s sign, abnormal Romberg’s sign, and instability of gait.
Cerebral MRI (23-10-2015): Bilateral frontal occipital parietal lobe, basal nerve nucleus region, corpus callosum, pontine multiple ischemic focus and infarction, lateral ventricular Para myelopathy, signs of brain atrophy.
Head MRI and MRI (December 11, 2015): Parabasal nerve nucleus showed long T1-length T2-signaling shadows, lateral ventricle, two lateral basal nerve nuclei, radiating corona, pontine, corpus callosum, and frontal parietal lobe showed multiple spot-thin T1-length T2-signaling shadows, FLAIR showed hypointense change, FLAIR showed spot-thin hyperintense signaling shadows in paraventricular medullary region. The anterior and middle cerebral arteries on both sides were locally narrow, hypointense, sparse distal to the artery, and the internal carotid and basilar arteries were tortuous.
Laboratory findings: Folate 4.24 ng/mL, low; cerebrospinal fluid protein 0.52 g/L, slightly high; conventional cerebrospinal fluid: white cell count 80x106 /L, mononuclear, 60%, multinuclear, 40%.
Case 2 underwent sequencing of NOTCH3 mutation in CADASIL disease (Shanghai Jindu Medical Examination Institute): Detected a heterozygous mutation: Zone: 19p13, reference sequence: NM-000435.2, location: Exon25, cDNA level: C.4699C>T, protein level: p. (Pro1567Ser) Diagnosis: CADASIL.
Examination: The right knee had a tendinous reflex and was positive for Chaddock’s sign on the right side, normal ataxia, rotation of the eyes to the left, and no cooperative Romberg’s sign on the left side. Normal gait with help.
Laboratory findings: Lipid level slightly elevated, lipoprotein-associated phospholipase A2 was 690.30 IU/L, and the rest were normal.
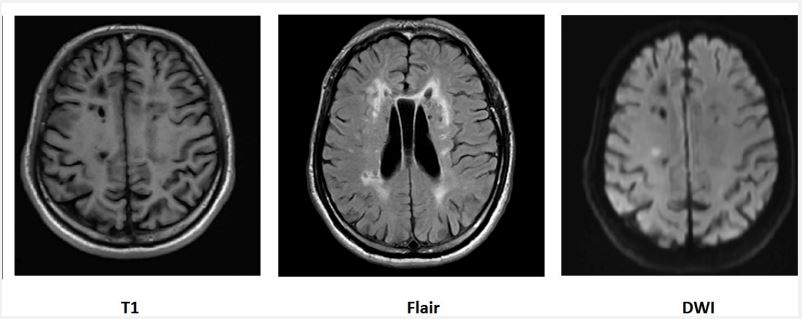
MRI of the head: Acute cerebral infarct in the right parietal lobe, multiple lacunae of softening and ischemia in the two basal nerve nuclei, corpus callosum, and front parietal occipital lobe, lateral ventricular para myelopathy (Figure 3); senile brain changes (high signal in corpus callosum, high signal in the outer capsule).
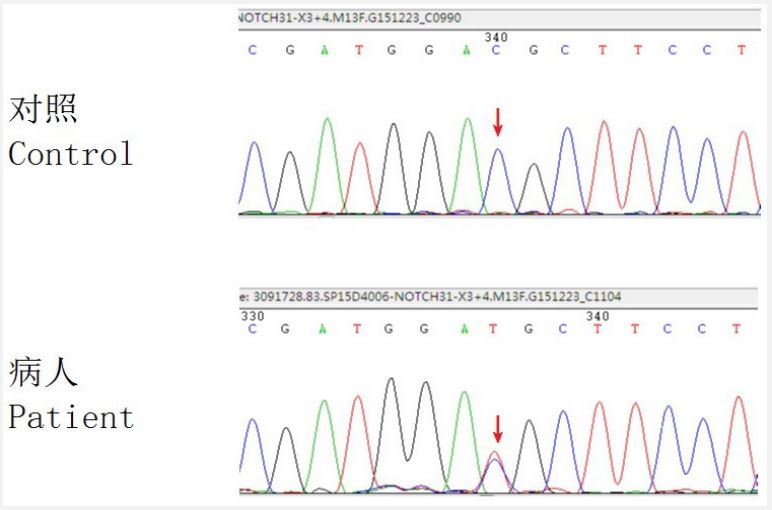
The cause of CADASIL was a mutation in the NOTCH3 gene expressed in vascular wall cells and capillary perivascular cells that resulted in mutations in the protein conformation leading to denaturation of molecular assembly. The 421C>T mutation detected in case 1, a common mutation site, was detected in 6.25% of all mutations in CADASIL [2]. The c.4699C>T mutation detected in case 2 (which is expected to convert Ser from amino acid 1567 of NOTCH3) was not reported by CADASIL and was not detected in the relevant gene databases (including 1000 genomes, oligonucleotide polymorphism databases, and exome sequencing program 6500siv2_ALL); PolyPhen-2 software speculated that the mutation was pathogenic [2,3]. Diagnosis by genetic sequencing is the primary diagnostic criterion. In recent years, a growing number of mutational sites have been identified, and although the relationship between these different sites and lesions has not been conclusively investigated, it is believed that this relationship will be revealed by more studies reporting on them [2,3]. Infarction is the most important pathological change in CADASIL, occurring predominantly in the medulla, less commonly in subcortical medulla, and newly discovered in the cortex. Infarction and subsequent dementia and psychiatric symptoms can be explained by knowledge of the anatomy and pathology as follows: Subcortical blood supply, including medulla, is in the central branch, subcortical blood supply is in the cortical branch, the subcortical branch is in the great middle artery, and the central branch is in the arterioles. This affects the arterioles and is thus a subcortical infarct. Because the central branch arises from the rougher arteries at a straight angle, it is itself an extremely susceptible site for cerebrovascular lesions, together with abnormal Notch 3 protein deposition and micro bleeds, and because of the micro vessel-specific lesions of CADASIL and the specific anatomy of the central branch, a wide range of cerebrovascular events occurs in the central area, which, in addition to the 100 percent incidence of the basal nerve nucleus, affects both the radiating corona of the internal and the external capsules, the latter being disease-characteristic. Of course, new findings suggest that infarction occurs in larger vessels, but this is not the main feature of the disease [2]. Characteristically, the imaging findings are fully present by age 35, but the typical clinical findings do not appear until after midlife [1,5]. It has been reported [5] that specific MRI findings in patients can be seen early in the course of the disease, but the diagnosis is often not made in time due to medical limitations, and there is a need for improved understanding of CADASIL. Several studies have shown that [3,6] CADASIL symptoms differ from those seen in Western countries in East Asian populations, with only 5%-10% of CADASIL patients experiencing migraine. But this does not mean that the migraine symptoms can be overlooked. 1. Migraine is the earliest symptom to appear by the age of 20 years, whereas other symptoms such as Transient Ischemic Attack (TIA) and stroke occur after the age of 40 years [1], and migraine is an indicator of early diagnosis.2. Migraine, particularly migraine with aura, is a major specificity of early CADASIL, and the study of migraine has important implications for the mechanism of CADASIL. The presence of migraine in the three members of case 1's family also suggests that migraine is a notable finding [2]. Bilateral small lesions distributed symmetrically across the BMCN and the semicircle were characteristic of CADASIL in the two probands and were characteristic of both the corpus callosum and the external cystic lesions. Six members of both families had a history of cerebral infarction, and both the father and the grandmother of the two probands had characteristic features of cerebral infarction and dementia. From these data, it can be seen that migraine, subcortical brain infarcts, and dementia regularly occur in different age groups are typical clinical features of CADASIL, and subcortical brain infarcts, particularly in the outer capsular region and corpus callosum, are more characteristic in midlife; transient ischemic attacks or ischemic strokes occur repeatedly and eventually lead to vascular infarct dementia, and the incidence of dementia is high in elderly family members, which is consistent with published reports.