1Department of Physical Chemistry, School of Chemistry, College of Science, University of Tehran, Tehran, Iran.
2Department of Physical Chemistry, Chemistry and Chemical Engineering Research Center of Iran, Tehran, Iran.
Mitra Ashouri* & Rohoullah Firouzi*
Email: m.ashouri@ut.ac.ir & rfirouzi@ccerci.ac.ir
Received : Aug 09, 2023 Accepted : Sep 01, 2023 Published : Sep 08, 2023 Archived : www.meddiscoveries.org
Papain-like protease (PLpro) is one of the most promising targets for anti-SARS-CoV drugs because it is an essential multifunctional cysteine protease for corona viral replication. In this study, an unbiased existing bioactive library containing 1916 FDA-approved drugs has virtually been screened using a new successful computational pipeline in the framework of the ensemble docking strategy to identify potential binding molecules of SARS-CoV-2 PLpro. Based on our protocol, 20 FDA-approved drugs were found to bind the target enzyme with significant affinities and good geometries, suggesting their potential to be utilized against the virus. Our computational studies identified IMATINIB, a well-characterized drug used in the treatment of lymphoblastic and chronic myeloid leukemia, as a potential inhibitor against PLpro. Subsequently, SIMEPREVIR, NALDEMEDINE, TUCATINIB, and some other FDAapproved drugs exhibit great affinities to the PLpro. These drugs are used in the treatment of several diseases such as cancer, schizophrenia, hypertension, hepatitis C, HIV infection, and AIDS showed high affinities in the screening. The high affinity of ligands was rationalized by the newly identified allosteric site named “BL2 groove” which is distal from the classical active triads. Occupying this groove may disrupt access to the catalytic site and affect the protein function. The best binding mode for these inhibitors and the efficacy of hydrogen bonds and hydrophobic interactions on inhibitory activities of ligands were also disclosed. Furthermore, our studies provide significant molecular insight into PLpro inhibition that could aid in the development of new drugs for COVID-19 treatment.
Keywords: COVID-19; Virtual Screening; Ensemble docking simulations; FDA-approved drugs; Drug discovery; Papain-like protease (PLpro).
For months, an ongoing pandemic caused by a single-stranded RNA virus (SARS-CoV-2) has been one of the major challenges facing the world. However, efforts are underway to keep up with the flood of coronavirus and no definitive cure has been reported to date. In general, the multiple stages of the SARSCoV-2 life cycle provide potential targets for drug therapy [1- 10].
To fight the current pandemic, major attempts have been made to target the SARS-CoV-2 spike protein, RdRp (RNA-dependent RNA polymerase), and cysteine proteases (Chemotrypsin-like protease or main protease (3CLpro or Mpro) and Papainlike protease (PLpro)). In contrast to others, there are very few potent inhibitors of PLpro with efficacy validated experimentally [11]. The PLpro encoded within non-structural proteins (nsps) during the viral life cycle is one of the most promising targets for anti-SARS-CoV drugs [12]. Available crystal structures of PLpro reveal that this functional protein is composed of three domains: (a) a classic catalytic triad cysteine cleavage domain (Cys111, His272, Asp286) (b) a Zn-binding domain (Cys189, Cys192, Cys224, and Cys226) and (c) ubiquitin domain (Asp164, Arg166, and Glu167). Besides these significant domains, distal from the cysteine active site, a newly identified binding pocket, “BL2 groove”, has been identified in recent articles (consisting of residues Leu162, Asp164, Arg166, Glu167, Pro247, Pro248, Tyr264, Gly266, Asn267, Tyr268, Gln269, Tyr273, and Pro299) [13-15]. Varieties of potent natural and synthetic compounds, from natural herbal ones or diverse antiviral drugs, are introduced as therapeutic compounds (e.g., Remdesivir designed for the Ebola virus, Lopinavir/Ritonavir designed for the HIV, chloroquine, and hydroxychloroquine designed for anti-malarial action, etc.) [16-20]. These compounds are found to be effective but their efficacy still remains disputed. In this way, exploring the repurposing of existing drugs and developing new drugs are both of considerable interest [21].
Computer-aided drug design has been a reliable technique in designing novel and potent therapeutics. The availability of multiple experimental protein structures has led to coping with the challenge of protein flexibility and induced fit at a lower computational cost via the ensemble docking technique. In this way, trial compounds are docked into binding sites of the protein ensemble and then ranked according to their strength of binding to identify initial hits for further experimental studies [22,23].
The dynamics of proteins over their conformational ensembles enable them to harness thermodynamic fluctuations for specific recognition of their targets, including small molecules. Different approaches have been designed to incorporate receptor flexibility through ensemble docking. A receptor ensemble can be constructed computationally (e.g., molecular dynamics simulations, phase space sampling methods, etc.) or by using available experimental structures of different co-crystallized protein-ligand structures. However, the main challenge is selecting a representative set of structures for an efficient docking simulation [24,25]. In this study, the pairwise RMSD matrix of general binding site residues for available X-ray structure of PLpro is calculated and the single-linkage hierarchical agglomerative clustering method based on the Ward Variance Minimization Algorithm was employed to detect representative protein structures [26,27].
The development of new antiviral drugs is time-consuming [28]. Therefore, to speed up the process, we used a new docking protocol in the framework of the ensemble docking strategy to identify SARS-CoV-2 PLpro inhibitors from an unbiased existing bioactive library containing 1916 FDA-approved drugs [29]. Since the safety of these drugs is well established and their efficacy can be quickly tested, drug repurposing could be an efficient approach to find potential inhibitors against COVID-19. The performance of the docking protocol has been evaluated in predicting the correct binding modes for the available crystal structures of PLpro-ligand complexes.
PLpro structure preparation
To create a conformational ensemble of PLpro, 45 PLpro structures in the Protein Data Bank (PDB) were selected [30]. All protein chains were characterized and analysed based on information which recorded in their PDB files (e.g. mutated/modified residues, missing atoms/residues, ligands information, resolution, alternate locations, etc.) (Table S1 in the Supporting Information). It is noteworthy that 12 protein-ligand complexes were covalently bonded to the thiol group of the Cys111 side chain in an irreversible manner. In addition, Cys/Ser mutation were observed in Cys111 catalytic residue in 16 protein-chain structures (6WRH_A, 6XG3_A, 6YVA_A, 7CJD_D, 7CJD_A, 7CJD_B, 7CJD_C, 7CJM_B, 7D47_A, 7D47_B, 7JIR_A, 7JIT_A, 7JIV_A, 7KOJ_A, 7KOK_A, 7KRX_A). The 87 monomeric chains were identified from the 45 PDB files of the PLpro, which 36 of the chains were discarded due to having the missing functionally important segments in the catalytic pocket of the PLpro (which includes a Cys111-His272-Asp286 catalytic triad).
The intersection of residues involved in the formation of β-sheets in the thumb and palm domains which contain 41 residues was used to drive the structural alignment process (Table S2). All PLpro monomeric chains were aligned on the Cα atoms of the selected residues of 6WRH chain A as a reference structure, which is a high-quality crystal structure of the PLpro with a resolution of 1.6 Å. The pairwise RMSD values for all above Cα atoms between every pair of the aligned protein structures of the PLpro did not exceed 0.48 Å. Even though we are all aware that the average nature of the RMSD measure does not capture local structural changes/dissimilarities between structures [25], this small amount of RMSD indicates the significant conformational stability of β-sheets (in the presence or absence of ligands) for use in alignment staging. In the following sections, to gauge conformational changes and structural differences between two protein structures, pairwise RMSD values between the individual the same pairs of residues (including all backbone and side-chain atoms) belonging to various pairs of the aligned protein structures were evaluated. This subject will be clarified more in the next sections.
Defining PLpro active site
The structural analysis of protein structures showed that 46 out of 51 protein chains were in complex with at least one ligand - from very small molecular fragments to very large peptidomimetic inhibitors. All the ligands were characterized by their approximated center and size which were defined by the centroid and the longest distance between two atoms of the ligands, respectively (Table S3). The distribution of ligands’ position in the protein structure based on their scaled approximate bounding sphere is shown in Figure 1a. To create a lucid and tangible 2D-representation of ligand distribution in the Cartesian space of protein structure, a principal component analysis (PCA) was done on the ligands’ centroid [31,32]. Figure 1b shows the projection of the ligands’ centroid distribution onto the subspace spanned by the first two principal components (PC1 and PC2) obtained from PCA. In Figure 1b, closed and overlapped circles occupy the same region on the protein surface. By considering Figures 1a and 1b, six separate regions are observed on the protein space, the most populated one (33 out of 46) is located at the same region that is known as the PLpro binding site - a cleft between the thumb and palm domains [13- 15]. Other sparsely distributed ligands should be discarded for the binding site analysis.
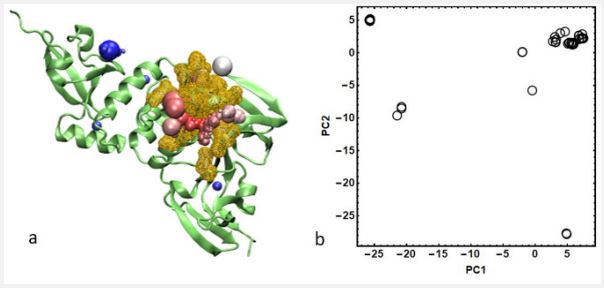
In order to define PLpro’s general active site, for all holo forms, all interacting amino acids that had even one heavy atom at a cutoff of 3.5 Å were included in the definition. Therefore, a pocket that was defined with 21 unique amino acids was considered as the general binding site of PLpro to use in further calculations (Table S4 and Figure 1a). This binding pocket definition is in good agreement with other studies based on different methods [15, 21]. It should be noticed that after defining the general active site, 6W9C chain A was omitted from protein structure data due to existing some missing residues in the active site.
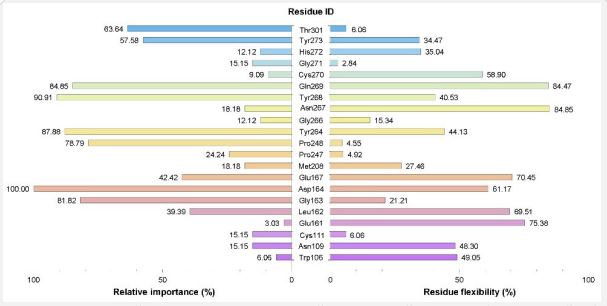
The “relative importance” of the general binding site residues was determined by the percentage of occurrence contacts of the residue in 33 protein-ligand complexes (see Table S4). For example, based on the “contact-importance” factor, Asp164 is the most important residue in PLpro active site. This amino acid is located at the ubiquitin domain of PLpro and plays a crucial role in protein activity [14,15]. Considering Table S4 shows that the general binding site includes all previous definitions of different PLpro domains (cysteine domain, ubiquitin domain, and BL2 groove) and is in good agreement with recent studies on PLpro active site [15, 33]. Relative importance data could be employed to select an optimal subset of PLpro residues for performing quantum mechanics (QM) calculations, especially for studying covalent inhibition mechanisms [6,34].
Another notable parameter for the active site residues is their flexibility. Accordingly, “residue flexibility” (RF) was defined as RF = 100*N𝑅𝑀𝑆𝐷/ (N2𝐶𝑜𝑚𝑝𝑙𝑒𝑥 − 𝑁𝐶𝑜𝑚𝑝𝑙𝑒𝑥) , where 𝑁𝑅𝑀𝑆𝐷 is the number of pairwise RMSD matrix elements with values more than 0.7 Å and 𝑁𝑐𝑜𝑚𝑝𝑙𝑒𝑥 is the number of complexes that RMSD calculation was done for their general binding site residues (3 complexes). In this way, the more flexible residues would have more RMSD matrix elements with values higher than 0.7 Å. The “relative importance” and the “residue flexibility” of the residues belonging to the general binding site have been displayed in Figure 2. For example, based on values reported in Figure 2 and Table S4 Asp164, Tyr268, Tyr264, Gln269, Gly163, and Pro248 residues in more than 75% of the complexes have effective contacts with their ligands (within the distance cutoff 3.5 Å). Consequently, such residues are important and should be considered for the determination of the binding pocket and this finding is in excellent agreement with recent studies [13-15]. Moreover, Thr301, Gly163, Asp164, Tyr264, Glu167, Gly271, and Pro242 have RF values lower than 60% and the loop structure. It did not seem farfetched that these most flexible residues belong to the BL2 groove. On the other hand, in comparison with other residues, Cys111 in the catalytic triad has the lowest flexibility. It is important to note that quantifying flexibility results can be used to identify and select an optimal set of flexible binding site residues for induced-fit docking (IFD) and flexible docking methods [35,36].
Conformational ensemble generation
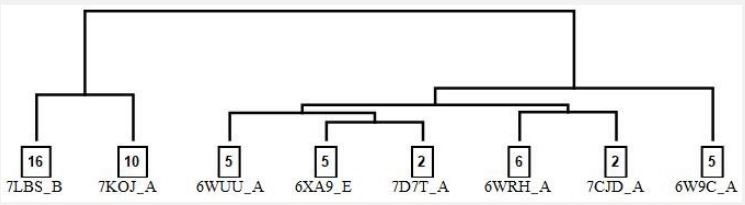
In this step, the pairwise RMSD matrix of general binding site residues was calculated for all 51 PLpro chains, and the singlelinkage hierarchical agglomerative clustering method based on the Ward Variance Minimization Algorithm was employed to detect representative protein structures [26,27]. A conformer belongs to a cluster if not only the pairwise RMSD average values with all members of the cluster are less than 1.2 Å but also the number of the pairwise RMSD values greater than 2.0 Å is less than three. The clustering result is depicted in Figure 3. As can be seen in this figure, the clustering yields 8 clusters that the population of each cluster is mentioned in the boxes. As it turns out, by loosening the clustering criteria clusters will merge and the number of them will decrease. Given the clustering constraints and the computational cost of calculations simultaneously, choosing eight conformers seems optimal for constructing a conformational ensemble of PLpro. In each cluster, structures with the lowest average RMSD than the others were selected as representatives. In cases where there were more than one representative conformer ones that had better resolution was chosen. PDB IDs and their corresponding chains for the representative structures were depicted in Figure 3.
Ensemble docking
For representative PLpro structures, hydrogen atoms, and missing atoms were retrieved using REDUCE software [37] and PSFGEN package within VMD software [38], respectively. During retrieving hydrogen atoms, side-chain flipping was considered based on the hydrogen-bonding patterns and the chemical environment, most of the histidine residues were protonated at Nε (i.e., HSE) and some of them were protonated at Nδ (e.g. His275). The prediction of protonation states and side-chain flips for the histidine residues found near the substrate-binding region can be found in Table S5. In addition, the basic residues (Arginine/Lysine) and the acidic residues (Aspartic/Glutamic) of all the structures were assigned positive and negative charges, respectively.
Protonated structures were optimized using the NAMD.2.13 program [39] with the CHARMM36m force field and generalized Born implicit solvent (GBIS) to regulate the structure in the CHARMM36m force field, and in particular to reduce the steric conflicts that may occur in the system. Minimization was performed for 20000 steps of the conjugated gradient where during the first 10000 steps protein heavy atoms were restrained with a positional harmonic force of 200 kcal/mol·Å2 followed by 10000 steps with the harmonic force of 100 kcal/mol·Å2. In all minimizations, the gradient has been reduced below 0.05 (kcal/mol)/Å.
Three-dimensional structures of the FDA-approved drugs, containing 1993 compounds, were downloaded from the Cheminformatic Tools and Database for Pharmacology site in the structure-data file (SDF) format [40]. The structures with more than 20 rotatable bonds were excluded from subsequent calculations, due to the well-known fact that the docking success rates decrease with increasing the number of active rotatable bonds [51,52]. In addition, six ligands that contain Boron element (B) were deleted from the ligands library due to the lack of force field parameters for this element. All 1910 remaining ligands were geometrically optimized in the gas phase using the PM7 [41] semi-empirical quantum mechanics (SQM) method with MOPAC2016 [42] while the gradient norm was set to 10 kcal mol−1Å−1. It is noteworthy that the PM7 is a fast and successful SQM method that reliably describes various types of noncovalent interactions and some important chemical observations, such as the planarity of conjugated rings or molecular fragments [43].
The initial docking search space was determined based on the active site definition of PLpro – as described in section 2.2. This definition includes all the bound ligands from small-molecule fragments to large peptidomimetic inhibitors at the catalytic binding site. Then the box size was extended by 5 Å in each of the three dimensions to ensure that the docking search space is large enough for the ligands to rotate in [44, 45]. The final docking box was included in a box of 20 × 23 × 27 Å3 , centered on the mean of the geometric centers of all of the considered ligands.
Docking simulations were performed for all 1910 ligands and 8 representative protein conformers with both AutoDock4.2.5.1 and AutoDock Vina1.1.2 software [44, 45]. Input files of docking with AutoDock were prepared automatically with Python scripts provided by MGLTools [46] and a Lamarckian genetic algorithm with an initial population of 500 was repeated 200 times for each ligand-receptor complex, therefore 200 models are built in each run. A grid spacing of 0.375 Å and a distance-dependent function of the dielectric constant was used for the calculation of the energetic maps with the program AutoGrid4 [47]. The default settings were used for all other parameters. For Vina calculations, the exhaustiveness parameter was increased to 500 in order to enhance the probability of finding the proper ligand conformations [48]. Each Vina run generates 20 poses.
All optimized ligands (1910 ligands) were docked in the conformational ensemble of the 8 prepared representatives PLpro structures (described in Sections 2.3). In total, 1600 (= 8 × 200) poses by AutoDock and 160 (= 8 × 20) poses by Vina were calculated for each ligand. All the predicted docking poses for each ligand were collected separately for each docking program and re-clustered based on the symmetry-corrected heavy-atom RMSD algorithm implemented in AutoDock4 with an RMSD cutoff of 2.0 Å [29, 53-57]. As a result, all predicted poses of a given ligand into multiple different conformations of the PLpro binding site are simultaneously organized and clustered for identifying representative poses and subsequent analyses. It is noteworthy to mention that using an ensemble of rigid receptor conformations in docking simulations (i.e., ensemble docking) is the most common strategy to cope with the receptor flexibility problem in rigid docking procedures [23,29,49,50]. Additionally, ligands flexibility are considered based on active rotatable bonds during the docking calculations, despite the fact that the docking success rates decrease with increasing the number of active rotatable bonds [51, 52].
Based on our recent researches, the top-ranked poses (with the lowest-energy poses) from the first and the most populated clusters are chosen as the representative poses for each docked ligand [29,43,53]. Therefore, we focus on the two representative docking poses for each ligand and use their docking scores, instead of considering the mean over docking score of the topranked poses of all representative PLpro conformations or taking the best-scoring binding poses from an aggregation of all predicted docking poses (the lowest scoring poses of ensemble docking), as routinely utilized in an ensemble docking protocol [23,49,50,55,56].
The above-mentioned computational pipeline (including the structural clustering strategy to construct the protein ensemble for performing docking calculations and the proposed manner to choose the representative docking poses from the ensemble docking results) has already been used for correctly predicting experimental binding poses and affinity ranking of Mpro-ligand complexes [29] and thus can be utilized to properly produce a rank-ordered list of the FDA-approved drugs. Accordingly, two rank-ordered lists of the docked FDA-approved drugs have been produced for each of both AutoDock and Vina software, one for those called the first clusters and others for the most populated clusters. The top 20 FDA-approved drugs for both the first and the most populated clusters are shown in Table 1 (the top 100 compounds are given in Tables S6 and S7 in the Supporting Information).
The analysis and comparison of the screening results show that there are several common drugs among the top 100 ranked-ordered lists of the aforementioned protocols. Eventually, 20 common FDA-approved drugs were found in all tables obtained from the representative poses of the first clusters and the most populated clusters (Tables S6,S7). These 20 common drugs and their medical uses in the treatment of different diseases and their 2D-chemical structures are shown in Table 2 and Figure S1. From the computational viewpoint, it means that these FDA-approved drugs target the PLpro protein and can be considered potential antiviral drug candidates in the treatment of COVID-19.
| First clusters | Most populated clusters | |||
|---|---|---|---|---|
| AutoDock | Vina | AutoDock | Vina | |
| Rank | Drug Names | Drug Names | ||
| 1 | VENETOCLAX | CONIVAPTAN | PLERIXAFOR | CONIVAPTAN |
| 2 | CAPMATINIB | OLAPARIB | GENTAMICIN | PAZOPANIB |
| 3 | IMATINIB | LUMACAFTOR | LURASIDONE | TROVAFLOXACIN |
| 4 | ZAFIRLUKAST | NILOTINIB | THIOTHIXENE | VEMURAFENIB |
| 5 | PLERIXAFOR | PAZOPANIB | PONATINIB | TELMISARTA |
| 6 | CABOZANTINIB | TROVAFLOXACIN | M RIFAPENTINE | PHENYTOIN |
| 7 | IRINOTECAN | VEMURAFENIB | PIMECROLIMUS | DOLUTEGRAVIR |
| 8 | GENTAMICIN | TELMISARTAN | ERIBULIN | ADAPALENE |
| 9 | LURASIDONE | PHENYTOIN | BRIGATINIB | BEXAROTENE |
| 10 | SIMEPREVIR | DOLUTEGRAVIR | TUCATINIB | TIPRANAVIR |
| 11 | THIOTHIXENE | BICTEGRAVIR | NETARSUDIL | MAZINDOL |
| 12 | PONATINIB | ENTRECTINIB | ZANUBRUTINIB | PERAMPANEL |
| 13 | M RIFAPENTINE | ADAPALENE | CALCIFEDIOL | OLAPARIB |
| 14 | PANCURONIUM | TROGLITAZONE | CALCITRIOL | IMATINIB |
| 15 | NALDEMEDINE | BEXAROTENE | SIMEPREVIR | LUMACAFTOR |
| 16 | TUCATINIB | TIPRANAVIR | IVERMECTIN | NETARSUDIL |
| 17 | EVEROLIMUS | MAZINDOL | TIPRANAVIR | FEXOFENADINE |
| 18 | CILOSTAZOL | CARBENICILLIN INDANYL | IMATINIB | NILOTINIB |
| 19 | PIMECROLIMUS | TROGLITAZONE | PIPERACETAZINE | GLYBURIDE |
| 20 | ERDAFITINIB | PERAMPANEL | RALOXIFENE | BINIMETINIB |
| No. | Name | First clusters | Most populated clusters | Used in the treatment of: | ||
|---|---|---|---|---|---|---|
| AutoDock | Vina | AutoDock | Vina | |||
| 1 | IMATINIB | -11.95 | -9.30 | -10.54 | -9.30 | Lymphoblastic and Chronic leukemia |
| 2 | SIMEPREVIR | -11.20 | -8.80 | -10.59 | -8.80 | Chronic Viral Hepatitis C |
| 3 | NALDEMEDINE | -11.07 | -9.00 | -9.61 | -8.50 | Constipation |
| 4 | TUCATINIB | -11.07 | -9.30 | -10.71 | -9.10 | Breast Cancer |
| 5 | ERDAFITINIB | -10.94 | -8.80 | -10.41 | -8.80 | Metastatic Urothelial Carcinoma |
| 6 | BRIGATINIB | -10.81 | -8.90 | -10.81 | -8.50 | Lung Cancer |
| 7 | NILOTINIB | -10.80 | -9.70 | -10.17 | -9.20 | Chronic Myelogenous Leukemia |
| 8 | ELTROMBOPAG | -10.76 | -9.10 | -10.08 | -9.10 | Cancer |
| 9 | TELMISARTAN | -10.70 | -9.60 | -10.12 | -9.60 | Hypertension |
| 10 | NETARSUDIL | -10.66 | -9.30 | -10.66 | -9.30 | Glaucoma or Ocular Hypertension |
| 11 | ZANUBRUTINIB | -10.64 | -9.40 | -10.64 | -8.70 | Mantle Cell Lymphoma (MCL) |
| 12 | TIPRANAVIR | -10.56 | -9.50 | -10.56 | -9.50 | HIV infection and AIDS |
| 13 | RISPERIDONE | -10.53 | -9.00 | -9.72 | -9.00 | Mania and Schizophrenia |
| 14 | PALIPERIDONE | -10.48 | -9.00 | -9.85 | -9.00 | Schizophrenia |
| 15 | CERITINIB | -10.33 | -8.90 | -9.63 | -8.80 | Lung Cancer |
| 16 | LUMACAFTOR | -10.31 | -9.80 | -9.93 | -9.30 | cystic fibrosis |
| 17 | AVAPRITINIB | -10.29 | -8.90 | -10.29 | -8.90 | Metastatic Gastrointestinal Stromal Tumours |
| 18 | MENTRECTINIB | -10.22 | -9.20 | -10.22 | -9.20 | Lung Cancer |
| 19 | BAZEDOXIFENE | -10.21 | -9.00 | -10.21 | -9.00 | Vasomotor Symptoms and Menopause |
| 20 | PERAMPANEL | -10.16 | -9.40 | -10.16 | -9.40 | Partial Onset Seizures |
The screening results show that a number of cancer drugs (IMATINIB, TUCATINIB, ERDAFITINIB, BRIGATINIB, NILOTINIB, ELTROMBOPAG, ZANUBRUTINIB, CERITINIB, AVAPRITINIB, MENTRECTINIB), schizophrenia drugs (RISPERIDONE, PALIPERIDONE), hypertension drugs (TELMISARTAN, NETARSUDIL), hepatitis C drug (SIMEPREVIR) and HIV infection and AIDS drug (TIPRANAVIR) display high binding affinities to PLpro protein. Our results are in agreement with others’ work that introduce possible drug candidates as PLpro inhibitors (e.g. Antivirus, Antihypertensive, Antibacterial, Antipsychotic, Anti-tumor, and anti-hepatic drugs) [58-61]. They demonstrate that some FDAapproved drugs that are used in the treatment of other diseases can be used as therapeutic agents in COVID-19. (e.g. Acetophenazine, Sulfasalazine, Dutasteride, Atovaquone). It is noteworthy that these drugs have also appeared in our 100 top-ranked lists.
A detailed survey of the chemical structures in Figure S1 demonstrates that most of the top-ranked compounds are Naphthalene-based that are of particular interest for their inhibitory activity against SARS-CoV-2 PLpro [60-63]. In some inhibitors, the Naphthyl ring is replaced with a biaryl group scaffold. It has been shown that this replacement is a promising development in this class of inhibitors as it likely represents a metabolic liability along the path to the clinic [64]. These findings can inspire the discovery of a new generation of naphthalene-based PLpro inhibitors.
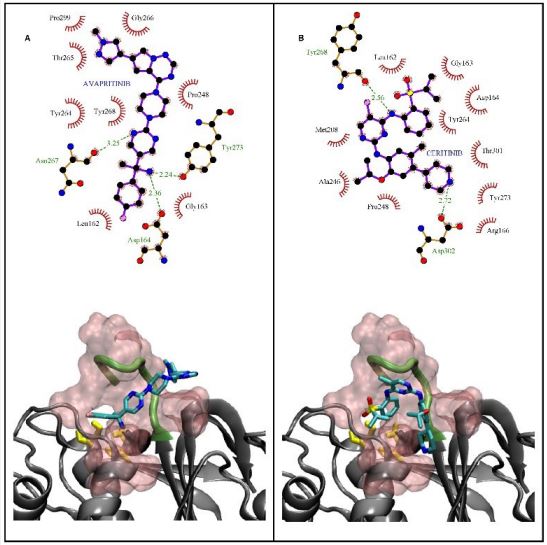
A detailed insight into the contacts and noncovalent interactions in docking-generated complexes has been obtained by LigPlot [65]. Careful inspection of the results shows that most of the 20 common ligands are located in the “BL2 Groove” binding pocket and Ubiquitin domain that are far from the classical catalytic triad. As mentioned before, the “BL2 Groove” is positioned at the N-terminal side of the BL2 loop and composed of hydrophobic amino acids such as Pro299, Pro248, Tyr264, Pro247, Gln269 and hydrogen bonding residues such as Gly266, Tyr268, Leu162, Asn267, Tyr273, Arg166, and Asp164. The strong hydrogen bonding and hydrophobic interaction between FDA-approved drugs with the enzyme imply that they can be potent PLpro inhibitors (Figure4 and Figure S2). Additionally, the inhibitors' orientations show that the BL2 loop is in direction of the catalytic site and occupying the BL2 groove can disrupt access to this site and affect the protein function by preventing the substrate from entering the active site in a non-competitive inhibition manner [60]. These results are in agreement with others' works and confirm the possibility of identifying allosteric inhibitors against SARS-CoV-2 PLpro [14, 66].
The SARS-CoV-2 papain-like protease (PLpro), is a key target for the design of inhibitors to tackle virus activity in host cells. Lately, computer-aided drug design techniques are reliable, affordable, and less time-consuming in designing novel therapeutics.
The objective of this work is the identification of potential inhibitors of SARS-CoV-2 PLpro protease from FDA-approved drugs using molecular docking. In this work, we used a new docking protocol in the framework of the ensemble docking strategy to identify SARS-CoV-2 PLpro inhibitors from a library containing 1910 FDA-approved drugs against PLpro proteases. The computational pipeline and its reliability are discussed in our recent work for Mpro-ligand complexes and has now been employed to properly rank 1910 FDA-approved drugs during their screening against PLpro target [29]. In this way, 100 top-ranked of drugs for four different used protocol were introduced and 20 common compounds were analyzed in detail. It has been shown that the highly flexible BL2 loop can regulate the binding cavity accessibility and besides the catalytic triad, the “BL2 Groove” should be considered as an allosteric pocket.
We demonstrate that some FDA-approved drugs which are used in the treatment of cancer, schizophrenia, hypertension, hepatitis C, HIV infection, and AIDS exhibit high binding affinity to PLpro protein and can be used as therapeutic candidates in the treatment of COVID-19. Obviously, performing comprehensive molecular dynamics (MD) simulations, experimental assays, and clinical trials are necessary to confirm their actual activity against COVID-19.
From a chemical scaffold point of view, we show that naphthalene-based drugs along with biaryl group substituted compounds have inhibitory activity against SARS-CoV-2 PLpro. Ensemble docking studies accented to the non-competitive inhibition mechanism and confirmed the existence of allosteric sites in the PLpro structure. The binding mode analysis confirms the contribution of hydrogen bonds along with extensive hydrophobic interactions to the inhibition of activity.
Conflicts of interest statement: There are no conflicts of interest to declare.
Data and software availability: MOPAC package and AutoDock Vina (version 1.1.2) were used under a free academic license for ligands preparation and docking simulations. Produced and analyzed data are available upon request.