1University-Affiliated Hospital, Qingdao University of Science and Technology, Qingdao, China.
2Liver Disease Center, the Affiliated Hospital of Qingdao University, Qingdao, China.
Fengchao Liu
Email: liangliang419519@163.com
Received : Jul 25, 2023 Accepted : Aug 14, 2023 Published : Aug 21, 2023 Archived : www.meddiscoveries.org
Hepatocellular Carcinoma (HCC) metastasis is the main cause of death in HCC patients, and clarifying the regulatory molecular mechanism of HCC metastatic progression will improve the therapies for HCC patients. The aim of this study was to identify hub genes and related signaling pathways involved in HCC metastasis. A total of 143 Differentially Expressed Genes (DEGs) were identified between primary and metastasis HCC samples using GSE40367 from the Gene Expression Omnibus (GEO) database. Gene Ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses of DEGs were performed for functional annotations, and a Protein-Protein Interaction (PPI) network was built. A total of 10 hub genes were identified from the PPI network, including GAPDH, H2AFZ, AFP, ALDOA, IGF2, AHSG, GART, FOS, APOA1, and HSPA8. The mRNA expression levels of the hub genes were significantly different between HCC and normal tissues. Kaplan‐Meier curves showed that GAPDH, H2AFZ, ALDOA, IGF2, AHSG, APOA1, and HSPA8 were closely associated with HCC patients’ overall survivals of based on the TCGA database. In addition, we established a prognostic model using Cox and LASSO regression in the TCGA cohort. A 3-gene signature (AFP, HSP18 and GAPDH) was constructed to stratify patients into two risk groups. Patients in the high-risk group showed significantly worse prognosis compared with patients in the low-risk group. In conclusion, the identified DEGs and signaling pathways may enhance our understanding of the molecular mechanisms of extrahepatic metastasis from primary HCC.
Keywords: Hepatocellular carcinoma; Metastasis; Hub genes; Differentially expressed genes; Bioinformatic analysis.
Hepatocellular Carcinoma (HCC) is one of the most prevalent malignancies and a leading cause of cancer-associated mortality worldwide [1]. Potential curative therapies include resection, liver transplantation, and local ablative therapies, but these are only available for early-stage patients [2]. Unfortunately, there are no effective treatment strategies for advanced-stage HCC, especially for patients with metastasis. Furthermore, even with early radical surgery, recurrence and metastasis are common and are responsible for the death of most patients [3]. Therefore, a deep understanding of the molecular mechanisms underlying HCC metastasis and the identification of precise anti-metastatic targets, are urgently needed to improve HCC prognosis.
In the past decade, systematic approaches for the analysis of whole tumor genomes and transcriptomes became affordable tools in tumor research [4]. Microarray-based technology and next-generation sequencing allow the examination of disease-related gene expression on a global genome scale [5]. Most studies focused on gene expression profiles of primary HCC tissues [6], but little is known about the molecular signature associated with metastatic HCC [7]. Analyzing differentially expressed genes between primary HCC and metastasis tissues may provide an important basis for understanding the mechanism of HCC metastasis.
To fulfill this purpose, we used bioinformatics approaches to identify DEGs between primary HCC and metastasis tissues based on the public Gene Expression Omnibus (GEO) datasets. Additionally, enrichment analyses including GO and KEGG were used to investigate the biological function of DEGs. Moreover, a Protein-Protein Interaction (PPI) network of these DEGs and hub genes were identified by the Cytoscape software. We analyzed the expression and prognosis of these key genes in HCC based on the TCGA database. Finally, we constructed a 3-gene signature on the basis of 10 hub genes, which was a good predictor of overall survival. Our study may clarify the underlying mechanisms of extrahepatic metastasis that are originated from primary HCC.
Microarray data
The transcriptome profiles of metastasis and HCC tissues were searched using GEO, a public data repository. We only included datasets that compared mRNA expression in HCC primary and metastatic HCC tissues. The gene expression profile dataset, GSE40367 [8], was extracted for further study. The dataset based on the platform of GPL570 (Affymetrix Human Genome U133 Plus 2.0 Array) contained 61 Samples. We extracted data from15 HCC primary tumor samples and 15 HCC metastasis tumor samples for further analysis.
Identification of DEGs
The DEGs between primary and metastasis samples were analyzed using GEO2R (https://www.ncbi.nlm.nih.gov/geo/ geo2r/), an interactive online analysis tool based on the R language limma package [9]. The DEGs of the dataset with an absolute log2 Fold Change (FC) > 1.5 and a p-value <0.05 were considered for subsequent analyses.
Gene ontology and KEGG pathway analysis of DEGs
Enrichment analysis of Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway for DEGs were performed using the clusterProfiler R package [10]. A p-value < 0.05 was considered statistically significant.
Protein–protein interaction (PPI) analysis and identification of hub genes
The interaction between these DEGs was analyzed using the Search Tool for the Retrieval of Interacting Genes (STRING, http://string.embl.de/), and a combined score of >0.4 was set as the cut-off criterion. Cytoscape software (version 3.8.2) was used to visualize the results of STRING database, and the degree algorithm in the cytoHubba plugin of the Cytoscape software was applied to identify hub genes [11]. We selected the top ten genes as hub genes for subsequent analyses.
Gene expression correlation analysis
Expression correlation analysis and visualization of hub genes based on TCGA samples was performed using R package “corrplot”. Gene Expression Profiling Interactive Analysis (GEPIA) was used to analyze differential expression patterns in different pathological stages of HCC [12].
Comparison of the expression levels of hub genes
To investigate the expression patterns of hub genes in HCC and non-tumor samples, the Fragments Per Million (FPKM) expression profile of 424 HCC samples (374 tumor and 50 nontumor) were retrieved from the TCGA database (https://cancer genome.nih.gov/). Statistical analysis and visualization of the expression of hub genes between HCC and non-tumor samples were implemented using R (version 3.6.3).
Survival analysis of hub genes
Kaplan-Meier overall survival analysis was used to evaluate the prognosis value of hub genes from 424 HCC patients in TCGA. According to the median gene expression levels, the patients were classified into high and low expression groups. The combination of Kaplan-Meier curve and log-rank test was used to test the survival difference between high and low expression groups using the R package “survminer” and “survival” [13].
Establishment of a prognostic signature model
To construct a prognostic risk signature, we conducted LASSO-COX analysis with the “glmnet” package to identify the most important hub genes for OS. The prognostic risk model was created by multivariate cox analysis. For evaluating HCC risk scores, the following formula was used: risk score =Σ (coef (hub gene) × expression value of hub gene). Patients were separated into high- or low-risk groups according to the median risk score, and survival difference between the two groups were compared by Kaplan–Meier curves. The performance of the model at different endpoints (1, 3, and 5 years) was then assessed via time dependent Receiver Operating Characteristic (ROC) curves using the R package “time ROC.” Multivariate Cox proportional hazards regression analysis was then used to determine if the risk score and clinical information were risk factors using the R package “survminer,” with a cutoff for risk factors of p<0.05. The nomogram was analyzed and depicted using the R package “nomogram.”[14].
Statistical analyses
Spearman was used to test the correlation between genes’ expression. The Wilcoxon signed-rank test was applied to analyze the difference in differentially expressed patterns of the hub genes. The survival difference between high and low gene expression groups was assessed by the log-rank test. A p<0.05 was considered statistically significant.
Identification of DEGs
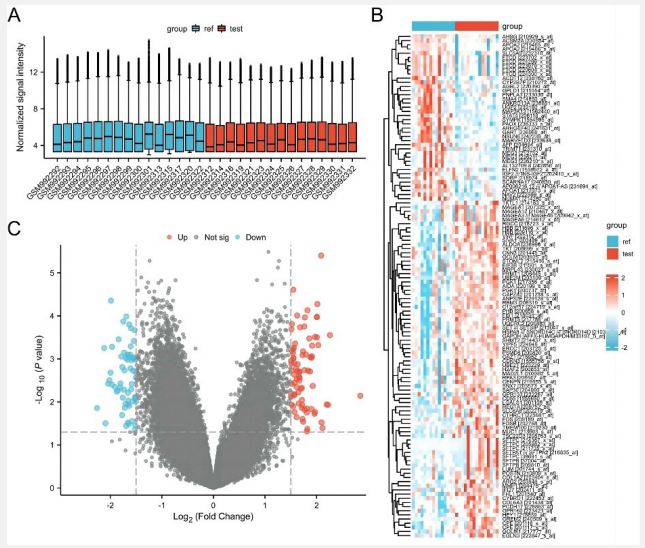
By comparing differentially expressed genes in the primary and metastatic HCC samples based on GSE40367 (Figure 1A), under the threshold of P-value < 0.05 and |logFC| > 1.5, a total of 143 DEGs (82 upregulated and 61 downregulated genes) were identified for subsequent analysis. A volcano plot and heat map of DEGs are shown in figure 1B and 1C, respectively.
Functional and pathway enrichment analyses of DEGs
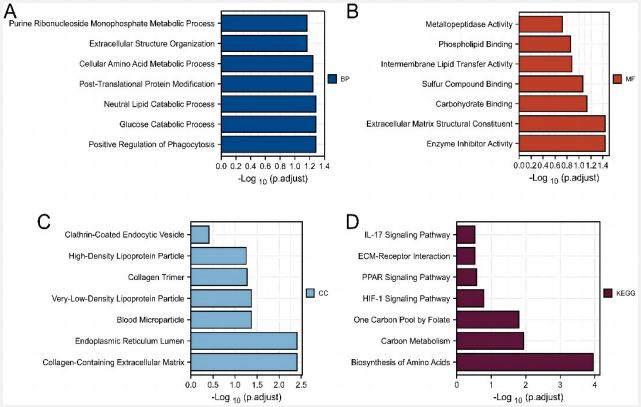
To investigate the function of these DEGs, we performed a GO analysis of the biological processes (BP), cellular components (CC), and molecular functions (MF). The BP analysis showed that these DEGs were significantly enriched in processes that regulate phagocytosis, glucose catabolism, lipid catabolism, post-translational protein modifications, amino acid metabolism, extracellular structure organization, and purine monophosphate metabolism (Figure 2A). For the MF analysis, the most enriched function was the enzyme inhibitory activity, extracellular matrix structural constituents, carbohydrate binding, sulfur compound binding, intermembrane lipid transfer activity, phospholipid binding, and metallopeptidase activity (Figure 2B). For the CC analysis, the DEGs were mainly enriched in collagen trimer, collagen-containing extracellular matrix, endoplasmic reticulum lumen, clathrin-coated endocytic vesicle, blood microparticle, and lipoprotein particle (Figure 2C). The results of the KEGG enrichment analysis showed that the most enriched pathways were biosynthesis of amino acids, carbon metabolism, one carbon pool by folate, the HIF-1 signaling pathway, the PPAR signaling pathway, ECM-receptor interaction, and the IL-17 signaling pathway (Figure 2D).
PPI network analysis and identification of hub genes
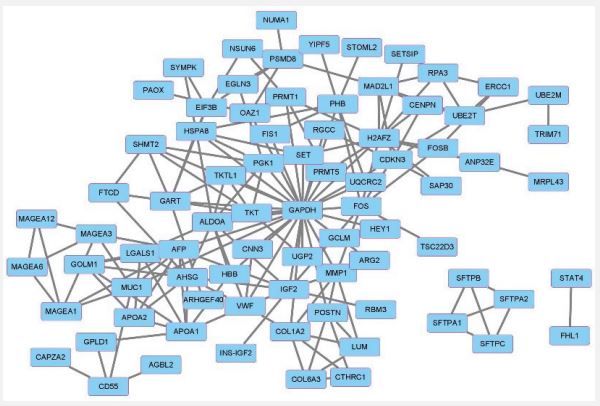
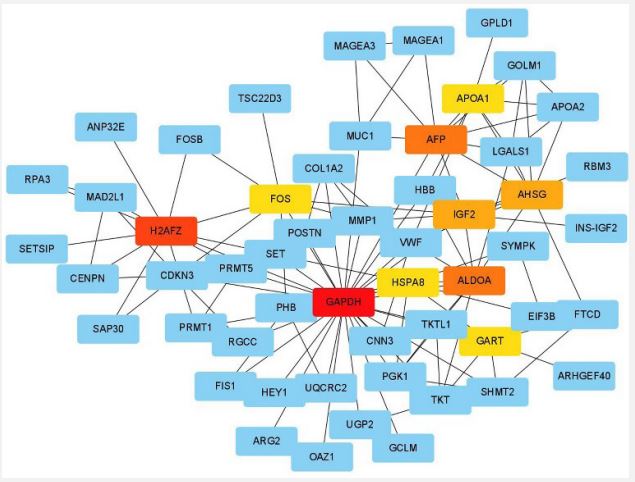
To explore the interaction between the DEGs, a PPI network consisting of 80 nodes and 169 edges was constructed (Figure 3). To trace the hub genes from the PPIs network, the Cytohubba plugin of the Cytoscape software was used. The hub genes were sorted according to their degree value to indicate the connectivity of the genes in the PPI network. The top 10 hub genes with a higher degree value were GAPDH, H2AFZ (H2AZ1), AFP, ALDOA, IGF2, AHSG, GART, FOS, APOA1, and HSPA8. The interactions between the Hub proteins and other proteins in the PPIs network are shown in Figure 4. The network consisted of 54 nodes and 123 edges.
PPIExpression patterns of the hub genes in HCC
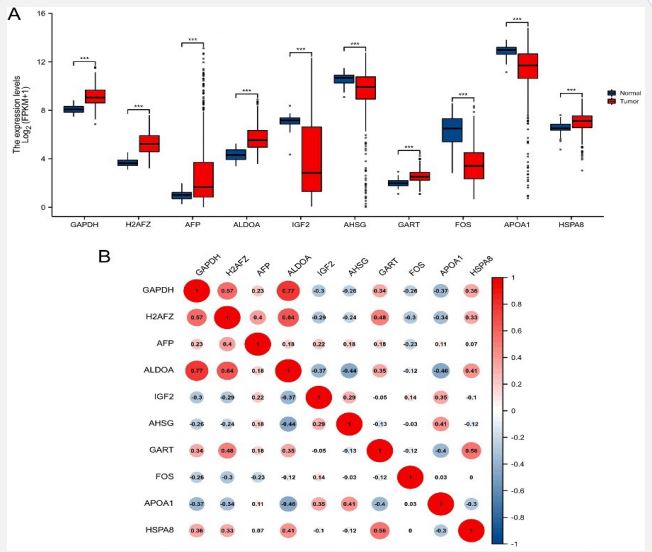
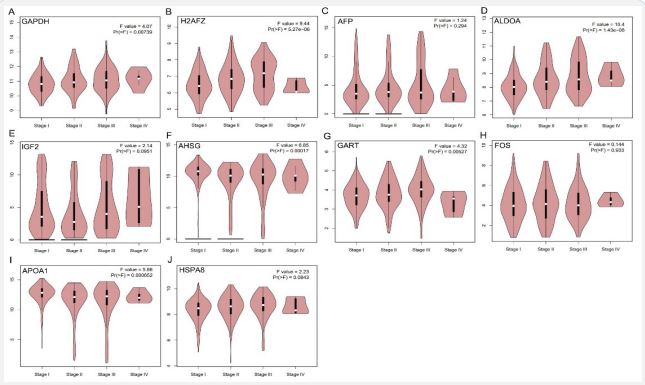
The expression levels of the hub genes in HCC tor normal liver tissues were evaluated using the TCGA database. The results showed that the mRNA expression levels of GAPDH, H2AFZ, AFP, ALDOA, GART, and HSPA8 were increased in HCC tissues when compared with those of normal tissues (Figure 5A). While IGF2, AHSG, FOS, and APOA1 mRNA expression levels were significantly decreased when compared with non-tumor tissues (Figure 5A). To investigate potential connections between the hub genes, we conducted a correlation analysis. The results showed a significant positive correlation between several hub genes, and a relationship between GAPDH and ALDOA which had the highest correlation score of 0.77 (Figure 5B). We then explored these genes expression patterns among different clinical stages of HCC, we found that except for the four genes AFP, IGF2, HSPA8, and FOS, all other hub genes in HCC tissues were differentially expressed among different pathological stages using the GEPIA database (Figure 6).
Survival analysis of the hub genes
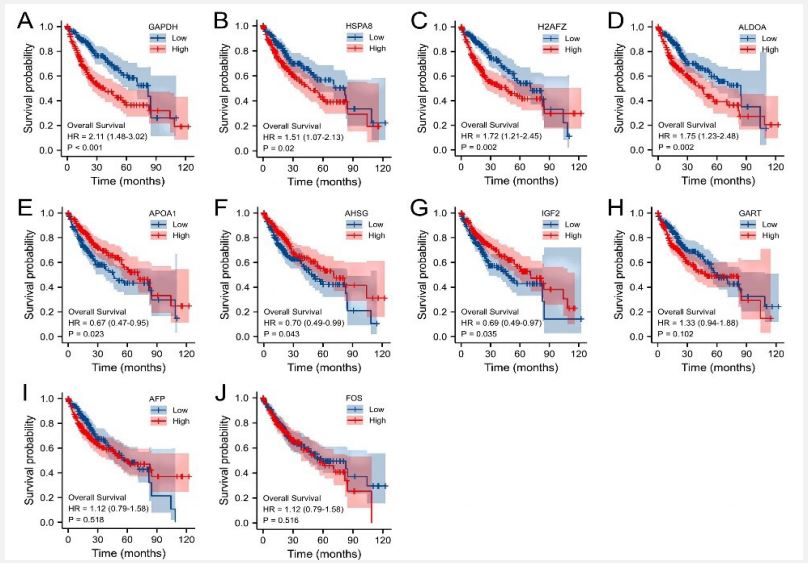
The prognosis value of the hub genes in HCC patients were evaluated using the TCGA database. To investigate whether the hub genes affect overall survival in HCC patients, we performed a Kaplan–Meier curve analysis. The results showed that high expressions of GAPDH, HSPA8, ALDOA, and H2AFZ were associated with worse overall survival (OS) in HCC patients (Figure 7A‐D). In contrast, APOA1, AHSG, and IGF2 were closely associated with better overall survival in HCC patients (Figure 7E-G). However, the expressions of GART, FOS, and AFP did not have significant influences on overall survival (Figure 7H‐J).
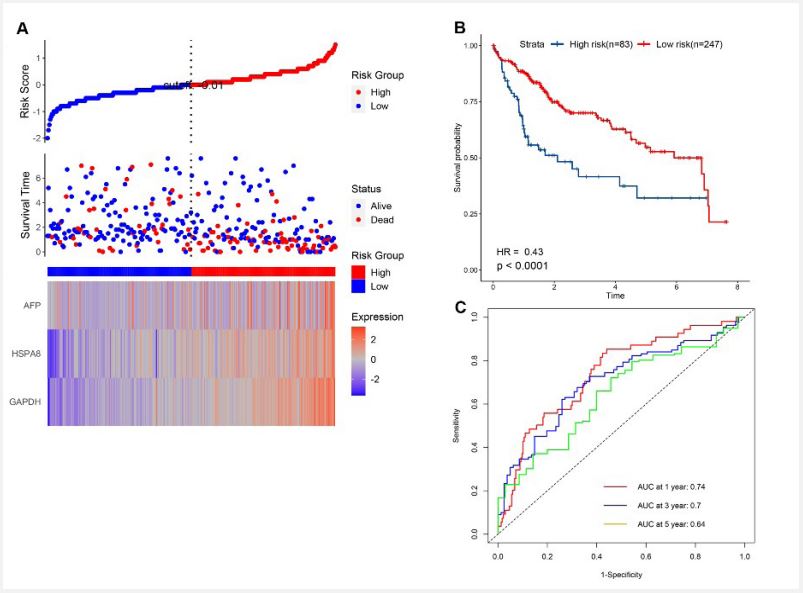
Construction of a prognostic model in the TCGA cohort
To establish a prognostic model, LASSO regression analysis was applied to identify 3 genes (AFP, HSP18 and GAPDH) from 10 hub genes. Then, we calculated the risk score of each HCC patient from TCGA and constructed a gene-signature based on these 3 genes. RS (risk score) = (0.3529 × expression of GAPDH) + (0.1792 × expression of HSPA8) + (0.0019 × expression of AFP). Subsequently, 330 HCC patients with follow-up information were divided into low-risk group and high-risk group according to the median value of risk scores in the TCGA (Figure 8A). The Kaplan–Meier survival curve was applied to show that patients in the high risk group had poorer outcomes than patients at the low risk group (Figure 8B). Besides, the time-dependent ROC curve was used to assess the prognostic ability of the 3-gene signature, and AUCs of the 3-gene signature at 1, 3, and 5 years were 0.74, 0.7 and 0.64, respectively (Figure 8C).
Metastasis is a multistage process that includes local invasion by the primary tumor cells, intravasation into the blood or lymphatic system, survival in circulation (hematogenous and/or lymphatic), arrest at a distant organ, extravasation, survival in a new environment, and metastatic colonization [15]. Studying the genetic differences between primary tumors and metastatic lesions may shed light on the mechanism of metastasis. With the development of microarray and sequencing technology, more studies compared the genetic differences between primary and metastatic tumors in a variety of solid tumors [16-19], including HCC [8] and a significant information are still missing. Therefore, bioinformatics analysis of the large data derived from these studies may help uncover hidden high-value information.
In this study, we systematically integrated a microarray dataset that contained primary and metastatic HCC samples. To have a better explore the potential mechanism of HCC metastasis, we first screened the DEGs in the two sets of samples, and then performed GO function and KEGG pathway analysis of these DEGs. Biological process terms of the GO analysis revealed that the DEGs are mainly enriched metabolic processes. Rewiring of the metabolic programs of glucose, amino acids and lipids is crucial for cancer cells strive in a primary tumor by overcoming nutrients and energy deficits, and eventually survive and form metastases [20]. For molecular function terms, in addition to the Extracellular Matrix (ECM) and metallopeptidase that are recognized to be involved in tumor metastasis, DEGs were enriched in molecular binding functions including carbohydrate, sulfur compound, and phospholipid bindings. Cell-cell interactions and cell adhesion that are mediated by integrins and the glycan-binding receptors, are key mediators of cancer progression via facilitating hallmarks of cancer, including immune evasion and metastatic dissemination [21]. The top GO terms according to the cellular component are collagen trimer, collagen-containing extracellular matrix, endoplasmic reticulum, endocytic, blood microparticle, and lipoprotein particles.
The results of enrichment analysis in KEGG pathways revealed an enrichment of these DEGs in the biosynthesis of amino acids, carbon metabolism, one carbon pool by folate, the HIF-1 signaling pathway, the PPAR signaling pathway, ECMreceptor interaction, and the IL-17 signaling pathway. Pathways including biosynthesis of amino acids, carbon metabolism, one carbon pool by folate also indicate that metabolic reprogramming may play an important role in HCC metastasis, which is consistent with the results of GO terms. Hypoxia is a potent microenvironmental factor that promotes metastatic progression [22]. Hypoxia and the expression of the hypoxia-inducible transcription factor HIF-1 are associated with increased distant metastasis and poor survival [23]. The Peroxisome ProliferatorActivated Receptor (PPAR) signaling has ambiguous functions in cancer development, and its relevance in advanced metastasis has been reported [24]. Membrane protrusion and adhesion to the Extracellular Matrix (ECM), which involves the extension of actin filaments and formation of adhesion complexes, are fundamental processes for metastasis [25]. IL-17 is a potent proinflammatory cytokine that contributes to HCC progression [26,27]; however, its role in the metastasis cascade of HCC remains largely unknown.
The hub genes were obtained through PPI network analysis and included GAPDH, H2AFZ, AFP, ALDOA, IGF2, AHSG, GART, FOS, APOA1, and HSPA8 genes due to their high degree values. The expression levels of the hub genes in HCC or normal liver tissues were evaluated using the TCGA database. The results showed that the mRNA expression levels of GAPDH, H2AFZ, AFP, ALDOA, GART, and HSPA8 were increased in HCC tissues compared with those of normal tissues. While IGF2, AHSG, FOS, and APOA1 mRNA expression levels were significantly decreased compared with those of non-tumor tissues. GAPDH, H2AFZ, ALDOA, and HSPA8 mRNA expression levels that were highly expressed in metastases were also highly expressed in HCC tissues when compared with normal liver tissues. IGF2, AHSG and APOA1 mRNA low expression levels in metastatic lesions were also expressed at low levels in HCC tissues when compared with normal liver tissues. Interestingly, the two genes AFP and GART that were highly expressed in metastases were expressed at low levels in liver cancer tissues compared with those in normal liver tissues. The FOS gene that was expressed at low level in metastases was highly expressed in liver cancer tissues. Surprisingly, except for the three genes AFP, GART, and FOS, the expression levels of all other hub genes in HCC tissues had significant evaluation values for the prognosis of HCC patients. Furthermore, we also analyzed the correlation between the expression levels of 10 hub genes and found that there was a significant positive correlation between GAPDH and ALDOA. These two genes have been reported to play an important role in the metabolic reprogramming of HCC [28,29].
In summary, the present study systematically integrated multiple transcriptomic analysis of HCC primary tumor site and metastatic tissues. We also identified DEGs and related signaling pathways that are involved in HCC metastasis. A total of 10 genes were identified as hub genes through PPI network analysis, and 7 genes showed prognosis-oriented values for HCC patients. We also constructed a 3-gene signature on the basis of 10 hub genes, which was a good predictor of overall survival. Collectively, our work provides novel insights into the mechanism of HCC metastasis and may support new ideas in therapeutic targeting and prognosis evaluation for HCC patients.
Funding: This work was supported by National Natural Science Foundation of China (82103662).
Acknowledgements: The authors would like to express their gratitude to EditSprings (https://www.editsprings.com/) for the expert linguistic services provided.
Author contributions: FCL conceived and designed the study. YZQ and FCL performed this study. FCL reviewed and edited the manuscript. All authors read, discussed and approved the final manuscript.
Conflict of interest: The authors declare that they have no conflict of interests.
Data availability statement: The dataset supporting the conclusions of this article is included within the article.