1University of Florida, College of Medicine, Gainesville, FL 32611, USA.
2Department of Neurosurgery, University of Florida, Gainesville, FL 32611, USA.
Ivelina P Kioutchoukova & Brandon Lucke-Wold
Email: ikioutchoukova@ufl.edu & brandon.lucke-wold@neurosurgery.ufl.edu
Received : Jul 04, 2023 Accepted : Aug 02, 2023 Published : Aug 09, 2023 Archived : www.meddiscoveries.org
Amyotrophic Lateral Sclerosis (ALS) is a severe neurodegenerative disease affecting the motor neurons. Although the etiology remains unknown, mutations in superoxide dismutase 1 have been observed in patients with familial ALS, resulting in increased calcium in the cells and leading to cell death. Additionally, studies in patients with the C9orf72 repeat expansion have shown lower age of onset, cognitive and behavioral impairments, and reduced survival. Accumulation of TDP-43 in the cytoplasm of neurons and glial cells caused by the loss of UBQLN2 has been shown to lead to mitotoxicity and proteasomal overload. Early diagnosis of ALS is necessary for the optimization of care between a patient’s neurologist and interdisciplinary team members to ensure the best outcomes possible. Proper management between physical therapy, occupation therapy, and pharmaceutical medications can improve ALS symptoms, achieving the highest quality of life possible for the patient. The current therapeutic medication recommended for ALS is Riluzole, but new therapies are emerging. This paper analyzes mechanisms of injury and progression of ALS along while analyzing current, emerging, and alternative therapeutics targeting ALS.
Keywords: ALS; SOD1; C9orf72; Riluzole; Relyvrio; Tofersen; Diet.
Amyotrophic Lateral Sclerosis (ALS) is a severe neurodegenerative disease affecting the motor neurons. It is the most common adult motor neuron disorder with heritable and sporadic forms [1]. Patients affected with ALS experience muscle atrophy, spasticity, and paralysis, along with damage to motor neurons in the motor cortex, brainstem, and spinal cord [1]. Within 4-6 years, most patients experience failure of their respiratory muscles, resulting in death. Although there is no known etiology, multiple mechanisms are currently being explored. 90% of patients present with sporadic ALS, which has an unknown cause, while the remainder present with familial ALS, potentially linked to superoxide dismutase mutations and C9orf72 repeat expansions [1]. Localization of TAR DNA binding protein 43 (TDP-43) to the cytoplasm of cells has also been studied and might offer insight into the mechanism of ALS.
Mechanism of injury and progression of ALS
ALS causes injury and death of the lower motor neurons in the brainstem and spinal cord along with the upper motor neurons of the motor cortex, resulting in failure of the neuromuscular system, and leading to respiratory failure and death [2]. ALS has multiple patterns of onset, but most commonly begins with lower motor neuron degeneration in the proximal limbs [3]. Although ALS was initially considered a pure motor disease, some patients exhibit involvement of the spinocerebellar and sensory pathways [2]. Despite many potential mechanisms being investigated, a single etiology of ALS remains unknown.
Superoxide dismutase 1 (SOD1) mutations
One mechanism addresses a dominant inheritance pattern of familial ALS. In 20% of familial ALS, Superoxide Dismutase 1 (SOD1) is a common mutation [4]. Studies have found that mice that overexpress mutated SOD1 eventually develop progressive and fatal muscle weakness and atrophy caused by motor neuron degeneration [5,6]. Another study found that conformational changes mirroring mutant SOD1 in mice have been obtained from samples of the spinal cord in patients with sporadic ALS, suggesting that modifications to the wild-type SOD1 might play a role in the mechanism of pathogenicity [7]. This proposed mechanism also highlights that reactive microglia, astrocytes, and T cell are common characteristics of ALS, leading to inflammation and degeneration of motor neurons [8]. Microglia are resident macrophages in the nervous system and continuously monitor the environment for signs of infection or injury [9]. Upon activation, microglia secrete Tumor Necrosis Factor (TNF) α, resulting in increased production of oxidant molecules, like Nitric Oxide (NO) [8]. Microglia also serve an anti-inflammatory function by secreting Interleukin-4 (IL-4) and Interleukin-10 (IL10) to instigate repair and limit inflammation [8]. In ALS, studies have found that microglia that express mutated SOD1 release TNFα, inducing lipid peroxidation and protein carbonylation and impairing the function of glial cells and neurons [8]. Astrocytes regulate the concentration of extracellular neurotransmitters and maintain metabolic homeostasis in the nervous system [10]. In neurodegenerative diseases, such as ALS, mutant SOD1 is believed to sustain the neuroinflammatory response, which is hypothesized to contribute to neuronal damage [8]. Over 200 mutations have been documented in ALS patients which interfere with the motifs of SOD1, resulting in mutated proteins [11]. Mouse models illustrate that misfolded SOD1 proteins interrupt the binding of copper and zinc, disrupting intramolecular binding and protein dimerization [12]. Some studies suggest that mutations in SOD1 result in a release of toxic factors from astrocytes [8]. Astrocytes protect neurons from excitotoxicity, however, this mechanism is compromised in ALS patients and was also observed in experimental mouse models [13]. Loss of protection from excitotoxicity results in decreased expression of the glutamate transporter, EAAT2 [14]. Additionally, damage to neurons because of ALS reduces the expression of EAAT2, increasing the concentration of glutamate in the synaptic cleft [8]. Increased levels of glutamate lead to overstimulation of AMPA and NMDA receptors [15]. Calcium concentrations increase inside motor neurons, eventually leading to cell death [15]. One study found that transplanting glial-restricted precursor cells into mutant SOD1 mice demonstrates therapeutic benefits by restoring EAAT2 function [8]. Restoring the function of EAAT2 offers a promising route of treatment for ALS patients, however, further research is needed in humans and clinical trials to determine efficacy and safety.
C9orf72 repeat Expansion
In patients with ALS, around 50% develop cognitive impairment, with 1 in 7 developing frank Frontotemporal Dementia (FTD) [16]. Multiple data sources suggest that pure ALS, pure FTD, and ALS with FTD might exist on a spectrum, with pure ALS on one end and pure FTD on the other end [17]. Conventional linkage and genome-wide association studies have recognized an expanded hexanucleotide repeat in the C9orf72 gene [18,19]. The C9orf72 gene is believed to lead to a gain of toxicity from RNA repeats and increased levels of the C9orf72 protein [20]. One study has demonstrated that the C9orf72 complex has been shown to regulate cellular processes such as lysosome homeostasis, vesicle trafficking, and autophagy [20]. Additionally, in patients demonstrating C9orf72 mutations, neuroinflammation results [20]. Studies have shown that the C9orf72 repeat expansion was present in 60% of familial ALS and 10% of sporadic ALS cases [18,19]. Bryne et al.’s study shows that patients with the C9orf72 repeat expansion and ALS portrayed a recognizable subphenotype with a lower age of onset, familial history of neurodegeneration, cognitive and behavioral impairments, along with reduced survival [17]. These patients also demonstrated increased apathy and worsened executive functions [17]. However, further research is still needed to understand how the C9orf72 repeat expansion plays a direct role in the progression of ALS.
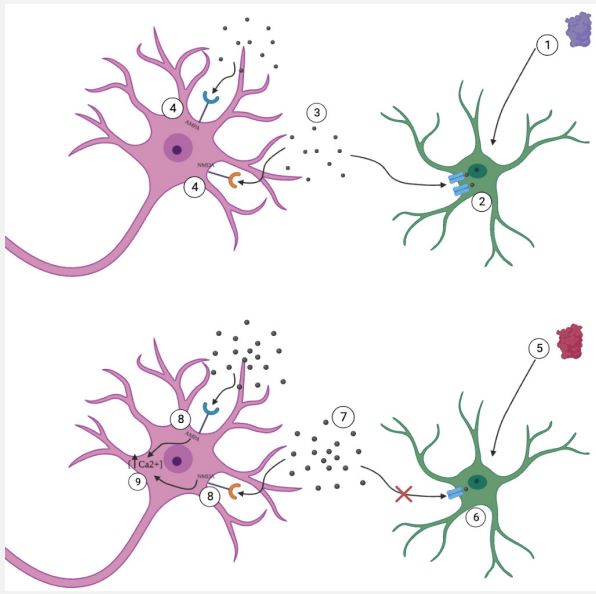
UBQLN2 levels
Protein aggregations disrupt normal protein homeostasis, resulting in cellular stress. In ALS, the cell is overwhelmed by the misfolded proteins and instead of using molecular chaperones to refold the proteins, ubiquitination or lysosomal degradation by autophagy occurs [21]. Ubiquilin-2 (UBQLN2) serves to deliver proteins tagged for ubiquitination to the proteasome [22]. Experiments have demonstrated that mice not expressing UBQLN2 demonstrated a phenotype consistent with human ALS features [23]. Loss of UBQLN2 function results in stress granule formation and TDP-43 localization in the cytoplasm [24]. In ALS, surviving neurons has been observed to have ubiquitin-positive inclusions [21]. Within these inclusions, the main component is TDP-43, an RNA and DNA-binding protein [25]. Localization of TDP-43 is a characterizing feature of ALS [26]. TDP-43 is involved in transcription, RNA transport, splicing, and stress granule formation [21]. Additionally, it shuttles between the nucleus and cytoplasm but primarily localizes in the nucleus [21]. However, in ALS, TDP-43 is localized to the cytoplasm [26]. One study showed that knockout mice without TDP-43 experienced early death, demonstrating that TDP-43 plays an essential role in normal development [27,28]. In sporadic ALS, abnormal cytoplasmic accumulation of TDP-43 in neurons and glia occurs in the motor cortex, brain stem, and spinal cord [26]. One study reported that TDP-43 accumulation resulted in mitotoxicity and proteasomal overload, but the sequence of events leading to neurodegeneration remains unknown [29]. Therefore, future research is needed to explore the role of TDP-43 through in vivo models, but it offers a potentially promising biomarker for diagnosis [26]. Additionally, further research is needed into the role of UBQLN2 levels on the progression of ALS, but it presents as a promising therapeutic target.
Diagnosis, management and long-term outcomes
ALS is the most common Motor Neuron Disease (MND) in adults and is a progressive disorder that often leads to devastating neurodegenerative outcomes. ALS affects 1-2.6 cases per 100,000 annually, with a median survival of 3-5 years [3]. The cumulative time-dependent survival at 1, 5, and 10 years from diagnosis was 76.2%, 23.4%, and 11.8%, respectively [30]. There is no biochemical marker found in serum or cerebrospinal fluid to establish the definitive diagnosis of ALS [31,32]. However, recent research suggests that using Neurofilaments (NF) and Electromyography (EMG) as biomarkers has the potential to improve ALS diagnosis. A meta-analysis of neurofilament use as an ALS biomarker suggests that NF heavy and light chains in the Cerebrospinal Fluid (CSF) of ALS patients were significantly elevated compared to healthy controls, NF light chains in the CSF were elevated in ALS patients compared to neurologic patients with conditions involving the Central Nervous System (CNS), and NF light chain concentration was higher in the blood of ALS patients compared to healthy controls [33,34]. It is believed that NF heavy and light chains can be used as a neural degeneration marker of ALS, but is not disease-specific, indicating that it may serve a better role in gauging disease progression [35]. A systematic review analyzing the use of EMG as a biomarker for ALS suggests that it may be a practical tool for analyzing patient data longitudinally to monitor disease progression [36]. Currently, the most validated method is the Motor Unit Number Index (MUNIX), which has only been implemented in two clinical trials [37]. While the use of EMG as a biomarker for ALS is promising, more research is needed to assess its use clinically.
Due to not having definitive diagnostic tests, the diagnosis of ALS is suggested through clinical examinations that indicate upper and lower motor neuron failure which often occur simultaneously [38]. Signs of Upper Motor Neuron (UMN) failure include muscle weakness, spasticity, hyperreflexia, and clonus. Lower Motor Neuron (LMN) findings include muscle atrophy and fasciculations [3,39]. The onset of ALS most commonly presents with LMN degeneration of the proximal limbs. This is characterized as Limb Onset (LO) ALS which makes up approximately 70% of ALS patients. LO ALS is characterized by LMN weakness and wasting and often present symmetrically in the proximal limbs before progressing distally, eventually severely impairing the function of the upper extremities and lower extremities. [40-42].
Due to the overlapping presentation of symptoms with other motor neuron diseases, ALS has the potential to go misdiagnosed. This delays the early referral to ALS specialty clinics and the use of drugs that would slow the rate of disease progression. There is no cure for ALS and early diagnosis and management of symptoms are essential for optimizing the patient’s quality of life [31,43,44].
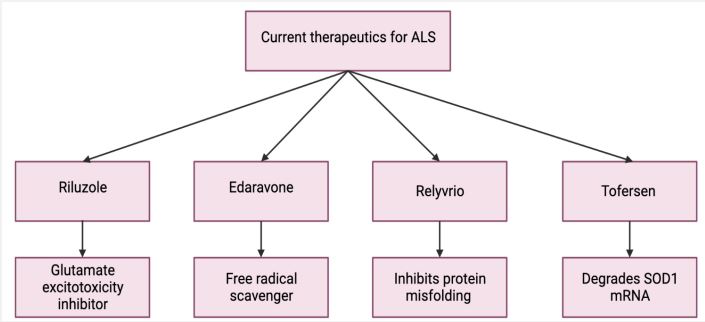
The current main treatment of ALS is symptom management and is a multidisciplinary effort between the patient’s neurologist, physical and occupational therapist, respiratory therapist, dietician, social workers, and nursing staff [45]. Physical therapy has been shown to slow down muscular deterioration in patients with ALS, allowing them to perform daily activities more optimally [46]. The two pharmacologic agents used for the management of ALS are Riluzole and Edaravone. Riluzole is suspected to reduce motor neuron damage through the inhibition of glutamate release, and Edaravone is suspected to serve as a neuroprotective agent by preventing oxidative stress by scavenging free radicals [47]. The most common cause of death in ALS patients is respiratory insufficiency which may be caused by various factors: diaphragmatic fatigue and weakness co-existing with respiratory muscle weakness causing reduced lung compliance and atelectasis, increased secretions increasing the risk of aspiration pneumonia, and bulbar dysfunction leading to nutritional deficiency causing fatigue of respiratory muscles. Management of these respiratory symptoms through non-invasive ventilation can increase enhance life quality and expectancy in ALS patients [48].
Riluzole, also known as 6-trifluoromethoxy-2-aminobenzothiazole, is the first FDA-approved pharmacological agent for ALS treatment [49,50]. Riluzole acts as a glutamate excitotoxicity inhibitor and provides cells with neuroprotective effects by preventing glutamate release from presynaptic neurons [50]. Additionally, Riluzole also suppresses the activation of sodium voltage-gated channels to further inhibit the glutamate excitotoxicity effect experienced in the neurons of ALS patients [50,51]. Riluzole is the only ALS treatment drug to have proven beneficial effects on ALS survival and demonstrate a slower progression of ALS [51,52]. To ensure the most benefits, ALS patients are instructed to take Riluzole long-term [52].
Edaravone is a new drug that has been approved by the FDA in 2017 to treat ALS. Edaravone serves as a free radical scavenger to reduce oxidative stress and has cytotoxic protective qualities to prevent degeneration of nerve cells [53]. One retrospective study testing the efficacy of Edaravone found the median survival of ALS patients provided with intravenous (IV) Edaravone was associated with a 6-month longer survival when compared to those in the control group [54]. This study found that the median survival for IV Edaravone treated cases was 29.5 months compared to non-IV Edaravone treated controls, which was 23.5 months [54]. However, in a study comparing the outcomes of ALS patients on Riluzole versus ALS patients on Riluzole with Edaravone, no significant difference was found in survival rates between groups, suggesting limited beneficial effects of Edavarone treatment [55]. Edaravone has limited clinical trials and mixed outcomes when testing its efficacy, thus suggesting further research and trials are needed on ALS patients [54].
A novel pharmacologic agent, more recently FDA-approved as of September 2022, called Relyvrio, demonstrates promising benefits to ALS patients [56]. Relyvrio consists of a combination of sodium phenyl butyrate and taurursodiol [57]. Relyvrio is thought to slow down the progression of ALS by inhibiting protein misfolding because of the endoplasmic reticulum stress and apoptotic response in the mitochondria of ALS patients [57]. In a randomized study testing Relyvrio treatment, statistical methods demonstrated a greater survival benefit of ALS patients treated with Relyvrio [58]. Despite these promising results, more clinical trials must be performed to confirm the efficacy of Relyvrio as a form of ALS treatment.
Tofersen is an FDA approved therapeutic for the treatment of ALS patients with the SOD1 mutation [59]. Tofersen is an antisense oligonucleotide that decreases SOD1 protein synthesis through the degradation of SOD1 mRNA [60]. One study demonstrated reduced concentrations of cerebrospinal fluid SOD1 and plasma NF light chains over 28 weeks. Tofersen was shown to have a positive impact on fast progressors. However, this did not improve clinical endpoints and adverse events were recorded following treatment [60]. Currently, research is being conducted in patients with presymptomatic familial ALS, which is characterized by an elevation in NF light chains, to investigate the efficacy of Tofersen [61].
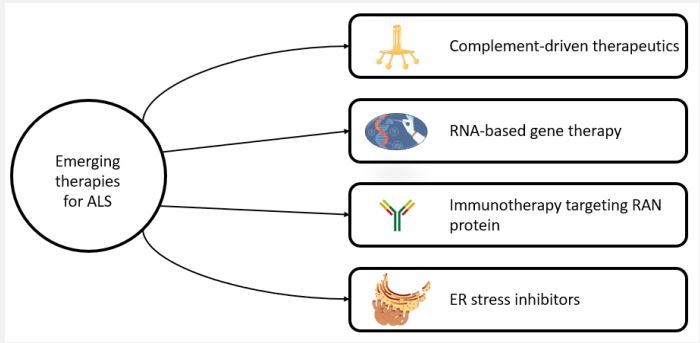
To this day, Riluzole is one of the only FDA-approved therapeutics that is being used to treat ALS. Despite that, multiple clinical trials have shown that it only extends survival by 2-3 months [62,63]. Thus, there is an urgent need to develop and research emerging and newly developing therapeutic agents for ALS.
Multiple studies have explored the possible involvement of the complement system using primarily animal models. For instance, one study performed an in-situ hybridization of ALS brains and spinal cords (post-mortem) and showed significant production of C1q and C4, components of the complement system [64]. Multiple other studies showed higher-than-normal levels of C3 and C4 in the serum of patients with ALS [65-67]. Some studies also showed an increased expression of C5a Receptor 1 (C5aR1) and reduced expression of Decay Accelerating Factor (DAF) and CD59, a membrane attack complex inhibitor [68,69]. Thus, complement-driven therapeutics are one of the crosswalks to consider while discussing emerging therapies for ALS. Two of the studies focused on the administration of PMX205, a C5 antagonist, in SOD1G93A mice. The results showed improved survival times along with reduced disease progression [70,71]. A similar study also demonstrated that deficiency in C5aR1 also improved survival rates in SOD1G93A mice [72]. However, deleting the gene encoding for C4 or C1q did not significantly affect the disease course [73,74]. Combining these results, it strongly identifies the C5a/C5aR1 as a target for ALS therapy.
In terms of a genetic cause, some research studies have discovered the C9orf72 repeat expansion as the most common cause of ALS [17,18]. Thus, targeting these expanded repeats has been a focus for many new therapies for this disease. One study approached this hypothesis by using miRNA, delivered via an Adeno-Associated Virus (AAV), that targeted the expansion repeats of interests in mice [75]. The results showed a decrease in C9orf72 expansion repeats. Moreover, a study showed positive results in SOD1-mediated ALS through AAV administration of miRNA in two patients [76]. Thus, reducing the expression of these mutated proteins is a promising therapy, however, it needs to be replicated and researched in a larger group of patients.
In addition to the expanded GGGGCC repeats observed in the non-coding region of C9orf72, other studies have found evidence pertaining to Repeat-Associated, Non-ATG (RAN) initiated dipeptide repeats that result from RAN translation (which include Dipeptide Repeat Protein (DRPs): Poly-GA, poly-GP, poly-GR, poly-PA, and poly-PR [77,78]. Thus, targeting RAN repeats has been one of the research areas for finding novel approaches. One study showed the production of α-GA antibodies in mice with GA-overexpression after vaccination with ovalbumin-(GA) 10 peptides [79]. These antibodies in turn prevented motor deficits by lowering the GA protein levels. Furthermore, in addition to lowering GA protein levels, this treatment also lowered GP and GR proteins, which is hypothesized to be a result of increased proteasome function.
Genetic studies investigating the disease pathogenesis of ALS have recognized that Endoplasmic Reticulum (ER) stress has also been demonstrated to play a significant role in the disease progression of ALS [80,81]. In terms of managing this etiology, some studies have used Salubrinal, a drug that reduces ER stress via activation of the Unfolded Protein Response (UPR) [82,83]. Moreover, mirroring this mechanism of action of Salubrinal in SOD1G93A mice increased survival rates and decreased the loss of muscle strength [84]. Another way to target ER stress is to upregulate a heat shock response through Heat shock factor 1 (Hsf1) which works by activating the chaperone protein gene expression and promoting the cellular protein-folding machinery. Arimoclomol, an inducer of heat shock response, has been found to decrease muscle degeneration in mice [85]. The efficacy of this drug in ALS patients is still being researched and is currently in the phase of a clinical trial [86]. Besides Arimoclomol, drugs like heat shock protein 90 (Hsp90) inhibitors (17AAG) that induce a similar heat shock response, may also be researched as part of novel therapies for ASL [87].
ALS patients often experience difficulties with swallowing, loss of appetite, and upper limb weakness, which hinder their ability to properly feed themselves and maintain proper nutrition [88]. Consequently, ALS is associated with weight loss and decreased muscle mass [89]. A shorter period of survival after diagnosis has been demonstrated with a faster decline in Body Mass Index (BMI) of ALS patients, thus emphasizing the importance of maintaining nutrition, energy intake, and BMI in ALS patients [89]. Therefore, one way to supplement treatment for ALS patients is to monitor their diet. Many diets have been suggested for ALS patients, such as high-calorie, ketogenic, antioxidant, high-fiber, vitamin-rich, and protein-heavy diets [88]. The goal of these diets is to improve gut health, prevent muscle loss, increase appetite, decrease the effects of oxidative stress, and slow the progression of ALS [88]. When specifically discussing vitamin E benefits, a study conducted by Veldink et al. suggests a 40-50% decline in ALS risk with the intake of vitamin E [90]. Additionally, when looking at high-fat diets, Fitzgerald et al. found a link between ω-3 polyunsaturated fats intake and decreased risk of ALS, further suggesting a positive and protective effect of high-fat diets on ALS survival [91]. Veldink et al. also suggest a 50-60% decline in ALS risk with the intake of polyunsaturated fatty acids, specifically omega-3 fatty acids [90]. Omega-3 fatty acids were reported as protective agents against glutamate excitotoxicity [89]. However, further research must be implemented to demonstrate the efficacy of these diets [88]. In addition to focusing on high-calorie diets, ALS patients are also recommended to stimulate their appetite with high-fiber diets and/or use cannabis, thus maintaining energy homeostasis [88]. Patients can also slow down ALS progression with enteral feeding and learning swallowing techniques [88].
Other alternate forms of ALS interventions include physical therapy, acupuncture, and stem cells. For ALS patients, physical therapists are instructed to perform staged and individualized exercise programs based on the ALS disease progression and autonomy of the patient [92]. As patients become more symptomatic and experience more impairments, physical therapists must adjust exercise programs and activity restrictions accordingly [92]. The goal of physical therapy seeks to maintain quality of life and mobility when possible as ALS progresses [92]. ALS patients tend to experience falls and respiratory issues as ALS progresses, thus physical therapists must instruct patients on exercises that promote respiratory muscle strength, as well as instruct patients on the use of assistive walking devices [92]. Despite the promotion of physical therapy to maintain the quality of life in ALS patients, physical exercise remains controversial as a form of intervention [92]. While stretching and working on range of motion is generally accepted for ALS patients, further studies must explore whether physical exercise beyond typical daily activities is beneficial to ALS patients [92]. Another form of alternative treatment includes acupuncture. Acupuncture is thought to inhibit glutamate excitotoxicity and slow down apoptosis of motor neurons [93]. However, further investigation must occur into the efficacy of acupuncture for ALS patients due to limited studies, mixed results, and varying study designs [93].
A newer form of intervention for ALS being explored includes the use of stem cells [94]. Stem cells, in theory, are thought to slow symptomatic loss of motor neuron function associated with ALS disease progression by differentiating into neuronal cells unaffected by the ALS and thus preventing neuron death where stem cells are placed [94]. Limited clinical trials and studies have been performed to determine the efficacy and safety of stem cell treatment for ALS patients [94]. Some animal studies in rats and mice demonstrated temporary improvement in motor function using stem cells [94]. However, given the design of the studies and with results only showing temporary improvement, the efficacy and safety of this intervention must be further investigated in future studies [94].
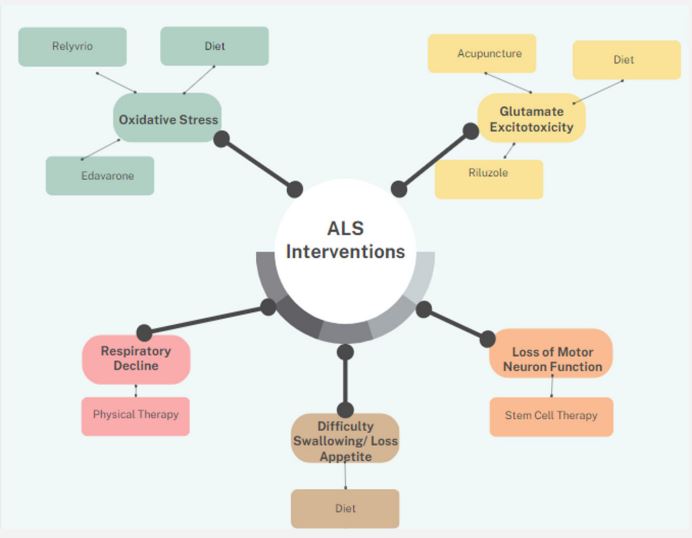
ALS is a progressive, neurodegenerative disease affecting both the upper and lower motor neurons. Although the etiology of sporadic ALS remains unknown, familial ALS may be influenced by mutations in SOD1 resulting in decreased expression of the glutamate receptor, EAAT2. This increases the levels of glutamate in the synaptic cleft, overstimulating AMPA and NMDA receptors which increase calcium levels in the cell, leading to cell death. C9orf72 repeat expansions have been linked to 60% of familial ALS, but further research is needed to elucidate its role in the progression of ALS. Additionally, loss of UBQLN2 expression results in localizations of TDP-43 in the cytoplasm, offering a potential biomarker for the diagnosis of ALS in patients. However, further research is needed through in vivo studies. Riluzole is thought to inhibit glutamate release, thus reducing motor neuron damage. Despite this, multiple clinical trials demonstrate it only extends survival by several months, demonstrating the need for new therapeutics to treat ALS. Several emerging therapeutics for ALS include Relyvrio, complement-driven therapeutics, miRNA targeting the C9orf72 repeat expansion, and Salubrinal. Alternative approaches also include vitamin E supplementation and omega-3 along with high-calorie diets. Implementing physical therapy in ALS patients may improve their quality of life. Although acupuncture and stem cell therapy provide new therapeutic options, further clinical trials and data are still needed to prove efficacy.
Author contributions: Conceptualization, B.L.-W, writingoriginal draft preparation, I.K., D.F., R.T., H.K, writing-review and editing, I.K., D.F., R.T., H.K., B.L.-W, supervision, B.L.-W.
Funding: This research received no external funding.
Institutional review board statement: Not applicable.
Informed consent statement: Not applicable.
Data availability statement: Not applicable.
Conflicts of interest: The authors declare no conflict of interest.