1Pharmaceutical Chemistry Department, Faculty of Pharmacy, Omar Almukhtar University, Al Bayda 991, Libya.
2Key Laboratory of Environmental Factors anChemistry Department, Faculty of Pharmacy, Heliopolis University for Sustainable Development, Cairo, Egypt.
3Pharmaceutical Organic Chemistry Department, Faculty of Pharmacy, Al-Azhar University, Nasr City 11884, Cairo, Egypt.
4Pharmaceutical Chemistry Department, Faculty of Pharmacy, Horus University - Egypt, International Costal Road, New Damietta,
Egypt
Khaled El-Adl
Email: khaled.eladl@hu.edu.eg
Received : Apr 03, 2023 Accepted : May 09, 2023 Published : May 16, 2023 Archived : www.meddiscoveries.org
The present study suggests the potential anti-SARS-CoV-2 activities of thirty five reported VEGFR-2 inhibitors containing the amide and urea linkers. Nineteen members revealed the best in silico results and hence, were subjected to further MD simulation for their inhibitory activities against SARS-CoV-2 Mpro across the 200 ns. Furthermore, Molecular Dynamics (MD) simulations followed by calculation the free energy of binding were also carried out for the most promising ligand-pocket complexes from docking studies to clarify some information on their dynamic and thermodynamic properties and approve the docking results. These results we obtained probably provided an excellent lead candidate for the development of therapeutic drugs against COVID-19. Both compounds Lenvatinib (4), Nintedanib (9), 28 and 32 as VEGFR-2 inhibitors bound to SARS-CoV-2 Mpro target have depicted significant free binding and affinity to target’s pocket. These results greatly confirmed our proposed rational and matched with our computational findings through molecular docking (targeting the viral Mpro) and molecular dynamics which consequently suggest the strong expected activities for the other studied and discussed VEGFR-2 inhibitors as well.
Keywords: Drug repurposing; Thiazolidine-2,4-dines; Docking; COVID-19; Main protease.
Drug repurposing/repositioning, a strategy effectively employed in cancer treatment [1], can represent a valid alternative, provided that suitable medications are selected among the enormous number of potential, already synthesized, and often already clinically employed, compounds. Drug repurposing has already been suggested for specific drugs in the treatment of the current COVID-19 outbreak [2-7]. Most of the drugs contemplated for repurposing/repositioning the treatment of the COVID-19 outbreak are commercially available, and their doses and toxicity in humans are well known, due to clinical use for years (or even decades). This can allow their utilization in faster and less expensive phase II–III clinical trials, or even within straightforward compassionate use. In particular, a significant number of drugs that have been reevaluated for COVID-19 treatment are or have been used as anticancer agents. This should not be surprising if we consider that virus-infected cells are pushed to enhance the synthesis of nucleic acids, protein and lipid, and boost their energy metabolism, in order to comply to the “viral program”. Indeed, the same features are seen in cancer cells, making it likely that drugs interfering with specific cancer cell pathways may be effective as well in defeating viral replication. In order to make a rational and effective choice of drugs amenable of repurposing for the therapy of COVID-19, we can elaborate existing data, from experimental and translational research, clinical trials, anecdotal reports and other published information. We present here a comprehensive analysis of available information on potential candidate VEGFR-2 inhibitors as cancer drugs that can be repurposed for the treatment of COVID-19.
There are four genera (α, β, γ, and δ) of corona viruses. Severe acute respiratory syndrome-related coronavirus (SARSCoV), the Middle East respiratory syndrome coronavirus (MERSCoV), and SARS-CoV-2 are β-coronaviruses [8]. Analysis of the genome sequences of these three viruses has revealed that SARS-CoV-2 has a higher identity to SARS-CoV (89.1% nucleotide similarity) than to MERS-CoV [9]. The genome of SARSCoV-2 is a positive-sense single-stranded RNA of approximately 30 kb in length and at least has six open reading frames (ORFs) that code for a minimum of 16 non-structural proteins and 4 structural proteins [10]. The 229E gene encodes two polyproteins involved in functional polypeptides release, which are required for viral transcription and replication. The proteolyticresponsible protease processing is 3 chymotrypsin-like proteases of SARS-CoV-2 (3CLpro or Mpro), as it cleaves at least 11 sites on the polyproteins translated from the viral RNA [11,12]. Given the relevance for the viral replication cycle, thus the viral protease (Mpro) has been proven as an attractive target in the development of inhibitors against coronaviruses [13-18].
Currently, there is no single specific antiviral therapy for COVID-19 and the main treatments is only supportive [19]. Drug repurposing is a strategy that adopted by several researchers to seek effective treatment in a short period [20]. Besides, the hypothetical assay based on molecular docking is emerging as an essential tool to discover new antiviral agents, as researchers can use this tool as a complementary approach prior to the synthesis of new compounds [21]. While traditional methods of drug discovery could take years, the approach taken here to search for possible medications for the SARS-CoV-2 is the in silico screening (molecular docking and dynamics simulations) of models towards the SARS-CoV-2 Mpro protein.
Rational of the work
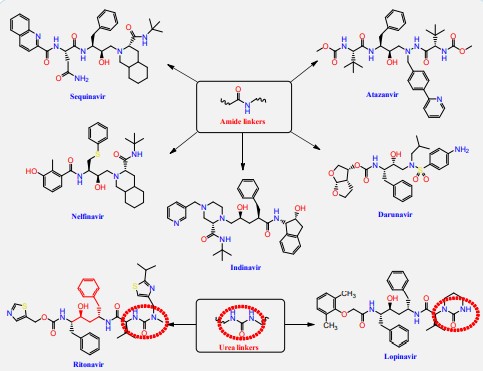
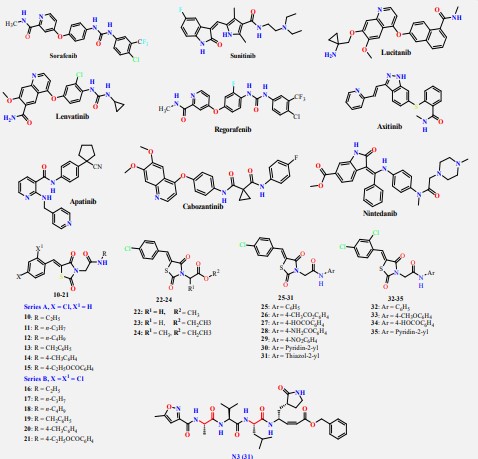
The HIV-1 protease inhibitor, nelfinavir, strongly inhibits the replication of the SARS coronavirus (SARS-CoV). Nelfinavir inhibits the cytopathic effect induced by SARS-CoV infection [22]. Similar to nelfinavir, ritonavir and lopinavir are recommended as protease inhibitors for the treatment of SARS and MERS, which have similar mechanisms of action as on HIV [23]. HIV-1 protease inhibitors on the one hand are structurally containing different amide and urea linkers (sequinavir, atazanvir, nelfinavir, fosamperavir, darunavir, indanavir, rotinavir and lopinavir) as depicted in Figure 1 [24].
Our rational depends on that VEGFR-2 inhibitors represent the main pharmacophoric urea and/or amide linkers (Figure 2).
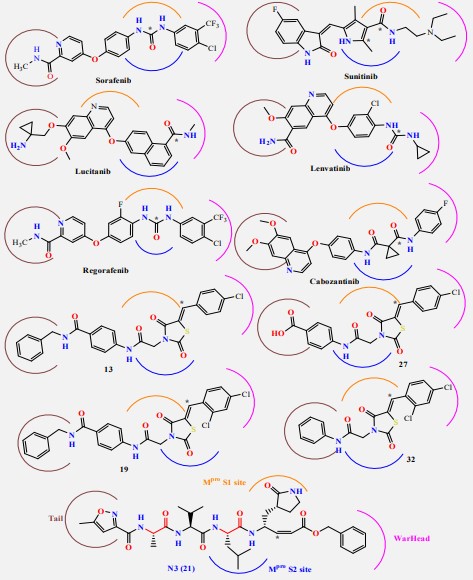
Our rational also depends on simulation of the inhibition process of SARSCoV-2 Mpro with N3. There are many N3-analogues as potential inhibitors, in which the recognition and warhead motifs are modified (Figures 2 & 3). QM/MM modeling of the mechanism of inhibition of Mpro by these compounds indicates that they may act as irreversible inhibitors and/or as a potential reversible inhibitor [25].
The best characterized Mpro inhibitors so far act with a covalent mechanism. They share a similar recognition moiety, i.e. a peptidomimetic scaffold of moderate size at the P1 position and a branched lipophilic group at P2 [26-28] and are equipped with a reactive warhead (Figures 2 & 3). Warheads so far employed for the design of SARS-CoV-2 inhibitors ranged from classical Michael acceptors (MAs) to activated carbonyl derivatives. Compounds equipped with less reactive warheads (i.e. carbonyl based compounds or nitriles) act as reversible inhibitors, as they form metastable adducts with cysteine residues. In terms of target engagement, duration of inhibition and efficacy, MAs have important potential advantages over other warheads [29].
In this research our derivatives are expected to be covalent inhibitors of SARS-CoV-2 as N3 specially thiazolidine2,4-dione derivatives and sunitinib were expected to be classical Michael acceptors (MAs).
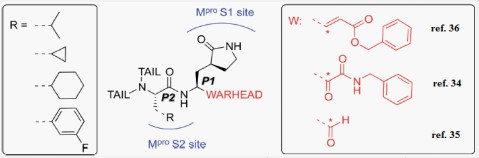
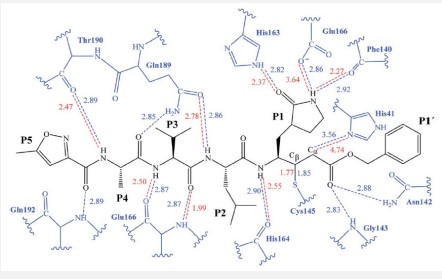
The first step of our program towards the design of new SARSCoV Mpro inhibitors was the study of the reaction with the N3 inhibitor originally proposed by Yang et al [30]. A schematic representation of the equilibrated structure of the active site is shown in Figure 4, where important interactions found in the MD simulations and the X-ray structure obtained by Jin et al [31] are indicated as blue and red dashed lines, respectively. The pattern of interactions between the enzyme and the inhibitor in our equilibrated structure is quite close to that observed crystallographically, thus supporting our starting structure for the exploration of the full mechanism. The MD results confirm the absence of hydrogen bond interactions with some of the side chains of the residues of N3 (P2–P5) which, considering the demonstrated efficiency of this inhibitor, can be used as a guide for the design of improved compounds not requiring hydrogen bond interactions with these sites as mentioned by Arafet et al [25].
On the other hand, the FDA-approved VEGFR-2 inhibitors (e.g. sorafinib containing urea linkers [32] and sunitinib containing amide linker [33]) and our studied and reported thiazolidine2,4-dione derivatives as VEGFR-2 inhibitors [34,35]) have structure similarity to HIV-1 protease inhibitors and/or the main pharmacophoric features of the co-crystallized inhibitor ligand of SARS-CoV-2 Mpro (N3) (Figure 5). So, the main objective of this study is to determine the efficiency of most VEGFR-2 inhibitors against SARS-CoV-2 using in silico docking and Molecular Dynamics (MD) simulation approaches. Besides, the studied VEGFR-2 inhibitors can be used as lead compounds for further optimization in the future based on SAR studied attaining better activity against SARS-CoV-2 Mpro.
Molecular docking studies
COVID-19 virus Mpro has a Cys–His catalytic dyad, and the substrate-binding site is located in a cleft between domain I and II. The N3 inhibitor is fitted inside the substrate-binding pocket of COVID-19 virus Mpro showing asymmetric unit containing only one polypeptide. Molecular docking simulation of VEGFR-2 inhibitors (1-35) and N3 inhibitor 36 into Mpro active site was done.
The docked results were compared to the crystal structure of the bound ligand–protein complex. The obtained success rates were highly excellent as cited in Table 1. The N3 ligand was docked into Mpro active site (pdb code: 6LU7). The RMSD of the docked ligand was 0.98 Å as it seems exactly superimposed on the native bound one (Figure 6). These results indicated the high accuracy of the docking simulation.
The proposed binding mode of N3 is highly matched with that obtained by Jin et al and exhibited eight H-bonds. It formed two H-bonds with GLU166 (1.80 Å and 1.94 Å), one H-bond with SER144 (1.70 Å), one H-bond with GLY143 (2.22 Å), one H-bond with PHE140 (2.43 Å), one H-bond with HIE164 (2.76 Å), one Hbond with GLN189 (2.33 Å) and one H-bond with THR190 (2.22 Å). Moreover, it was confirmed to form many hydrophobic interactions as it occupied different hydrophobic groves as shown in Figure 7.
| Ligand | Binding energy (kcal/mol) | Interacting amino acids | Distance Å | |
|---|---|---|---|---|
| Sorafenib | -8.0 | CYS 145 CYS 145 ASN 142 |
H-donor H-donor H-donor |
4.47 3.98 2.98 |
| Sunitinib | -7.5 | CYS 145 GLY 143 |
H-donor H-acceptor |
3.40 3.11 |
| Lucitanib | -8.0 | GLU 166 GLU 166 |
H-donor H-donor |
3.21 3.09 |
| Lenvatinib | -8.3 | ASN 142 | H-acceptor | 3.56 |
| Regorafenib | -7.5 | CYS 145 CYS 145 |
H-donor H-donor |
4.15 3.88 |
| Axitinib | -7.4 | ----- | ----- | --- |
| Apatinib | -7.5 | CYS 145 MET 165 MET 165 |
H-donor pi-H pi-H |
3.45 3.74 3.67 |
| Cabozantinib | -8.1 | MET 165 THR 190 GLU 166 THR 26 |
H-donor H-donor H-acceptor pi-H |
3.72 3.52 2.98 4.69 |
| Nintedanib | -7.7 | THR 26 ASN 142 |
H-donor H-acceptor |
3.34 2.98 |
| 10 | -7.2 | ASN 142 MET 165 CYS 145 |
H-donor H-donor H-acceptor |
3.49 3.66 3.08 |
| 11 | -7.1 | ASN 142 GLU 166 CYS 145 GLU 166 |
H-donor H-donor H-acceptor pi-H |
3.55 2.84 3.03 4.31 |
| 12 | -7.0 | ASN 142 GLU 166 CYS 145 GLU 166 |
H-donor H-donor H-acceptor pi-H |
3.39 2.94 3.02 4.27 |
| 13 | -8.1 | CYS 145 THR 25 |
H-donor H-acceptor |
3.34 2.98 |
| 14 | -7.4 | CYS 145 CYS 145 HIE 41 GLU 143 |
H-donor H- donor H-acceptor pi-H |
3.38 3.57 3.16 4.50 |
| 15 | -7.4 | GLN 189 GLY 143 GLU 166 |
H-donor H-acceptor H-acceptor |
3.01 2.94 2.95 |
| 16 | -7.4 | GLN 189 GLY 143 GLU 166 |
H-donor H-acceptor H-acceptor |
2.95 2.96 2.87 |
| 17 | -7.4 | HIP 164 THR 24 ASN 142 |
H-donor H-donor H-acceptor |
3.37 3.03 3.48 |
| 18 | -7.3 | ASN 142 CYS 145 GLU 166 |
H-donor H-donor H-acceptor |
3.53 2.95 4.17 |
| 19 | -8.1 | THR 26 ASN 119 TYR 118 |
H-donor H-donor pi-pi |
3.66 3.25 3.90 |
| 20 | -7.2 | ASN 142 CYS 145 GLU 166 |
H-donor H-acceptor pi-H |
3.56 3.07 4.31 |
| 21 | -7.3 | CYS 145 GLU 166 THR 25 |
H-donor H-donor pi-H |
4.43 3.52 4.14 |
| 22 | -6.2 | CYS 145 GLY 143 |
H- donor H-acceptor |
3.66 2.80 |
| 23 | -6.3 | GLY 143 SER 144 |
H- acceptor H-acceptor |
2.86 2.75 |
| 24 | -6.1 | ---- | ---- | ---- |
| 25 | -7.4 | CYS 145 CYS 145 CYS 145 HIE 41 |
H-donor H- donor H- donor H-acceptor |
3.31 3.63 3.97 3.06 |
| 26 | -7.4 | HIP 164 | H-donor | 2.98 |
| 27 | -7.0 | GLU 166 GLU 166 |
H-donor H-acceptor |
2.93 3.00 |
| 28 | -7.4 | LEU 141 ARG 188 THR 24 GLU 166 GLN 189 |
H-donor H-donor H-donor H-acceptor pi-H |
3.65 2.70 3.96 3.28 4.61 |
| 29 | -7.2 | LEU 141 THR 24 THR 190 GLU 166 GLN 189 |
H-donor H-donor H-donor H-acceptor pi-H |
3.66 3.92 2.97 3.31 4.61 |
| 30 | -7.3 | HIP 164 SER 46 |
H-donor H-acceptor |
3.38 2.96 |
| 31 | -7.2 | CYS 145 CYS 145 CYS 145 HIE 41 |
H-donor H-donor H-donor H-acceptor |
3.46 3.48 4.02 3.15 |
| 32 | -8.1 | CYS 145 CYS 145 CYS 145 HIE 41 THR 26 |
H-donor H-donor H-donor H-acceptor pi-H |
3.50 3.59 3.97 3.15 4.59 |
| 33 | -7.3 | LEU 141 THR 24 GLU 166 |
H-donor H-donor H-acceptor |
3.78 3.62 3.13 |
| 34 | -7.4 | LEU 141 GLU 166 HIE 41 THR 26 |
H-donor H-donor H-donor H-acceptor |
3.71 3.74 3.73 3.13 |
| 35 | -7.3 | LEU 141 GLN 189 THR 143 GLU 166 GLN 189 |
H-donor H-donor H-donor H-acceptor pi-H |
3.80 3.21 3.71 3.22 4.58 |
Finally, some VEGFR-2 inhibitors have excellent binding scores (-7.5 to -8.3) which are equipotent or better than the native ligand N3 (-8.1).
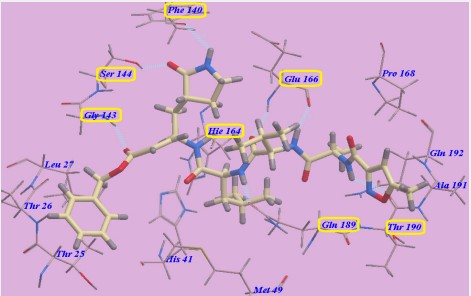
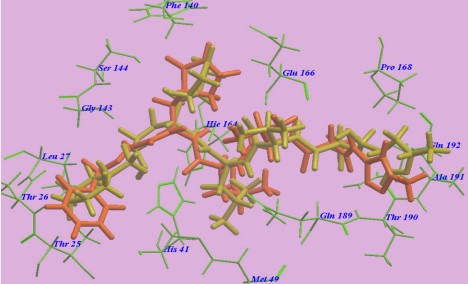
Most compounds showed nearly binding modes like the N3 inhibitor. Many poses were obtained with better binding modes and interactions inside the receptor pocket. The poses with the most acceptable scores (related to the stability of the pose) and rmsd_refine values (related to the closeness of the selected pose to the original ligand position inside the receptor pocket) were selected. The docked compounds got stabilized at the N3-binding site of Mpro by variable several electrostatic bonds (Table 2). Results of energies and different interactions with amino acids of Mpro protein pocket are shown in Table 1.
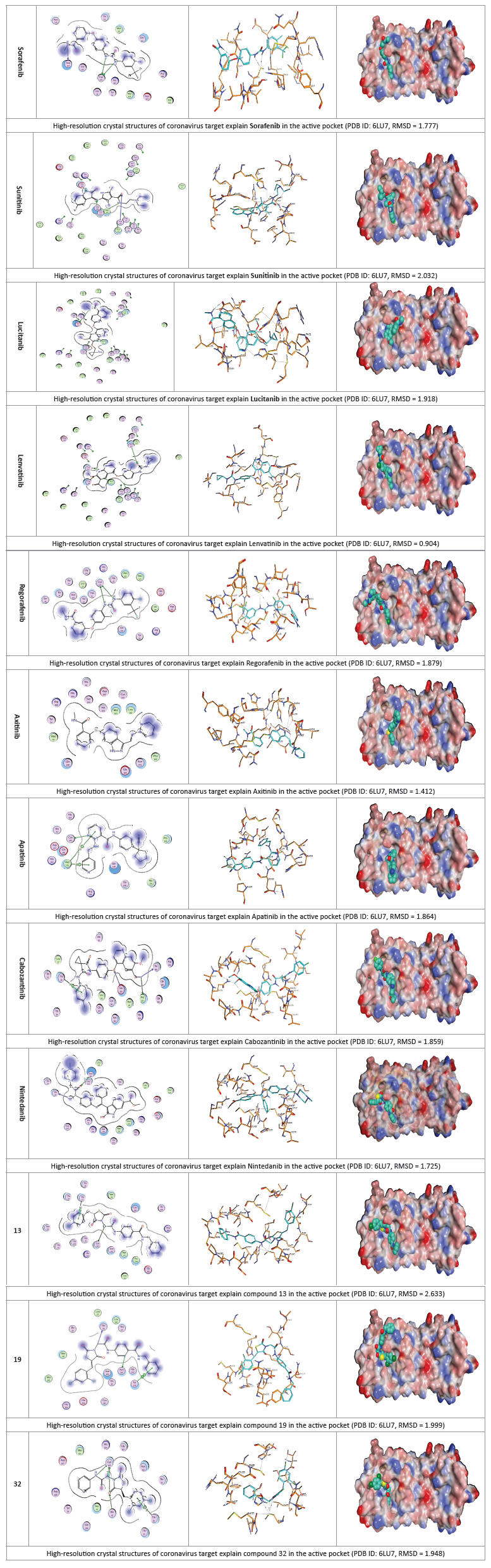
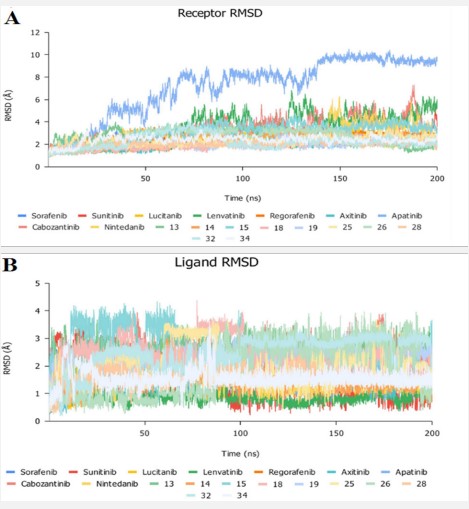
Binding-free energy calculations
The binding-free energy calculation was performed to understand the nature of ligand–protein interaction as well as obtain more detailed information concerning the individual ligand contribution [39]. In this regard, the molecular mechanics generalized Born surface area (MM/GBSA) calculation was implemented for binding-free energy estimation, where higher negative binding energy explains more ligand affinity towards its respective target pocket [40]. Utilizing implicit solvent models in MM/GBSA framework allows for efficient calculations without significant loss of accuracy. MM/GBSA demonstrated accurate pose prediction on a large benchmark of protein–ligand complexes with non-redundant binding poses [41]. Adopting the calculation across the 200 ns MD simulation time course was rationalized by the rapidly attained equilibration/convergence of the complex RMSD trajectories following few initial MD frames. To our delight, compounds Lenvatinib (4), Nintedanib (9), 28 and 32 as VEGFR-2 inhibitors bound to SARS-CoV-2 Mpro target have depicted significant free binding and affinity to target's pocket (Table 2) (Figure 10). Dissecting the obtained binding-free energy into its contributing energy terms, the van der Waal interactions showed superior contribution within the free-binding energy calculation of ligand–protein complex as compared to electrostatic potential energy except in case of sorafenib. Moreover, Gas-phase energy (ΔGgas) of the solute is electrostatic energies (ΔGgas = VDWAALS + EEL). The solvationfree energy, ΔGsolv, is broken into the polar and non-polar components ΔGsolv = ΔGpol + ΔGnonpol and then ΔTotal = ΔGgas + ∆Gsolv. N3 exhibited higher both ΔGgas and ΔGsolv compared to our VEGFR-2 inhibitors except in case of Lenvatinib, Nintedanib, compounds 28, and 32 which showed the highest ΔGsolv. Based on current literature, the Mpro pocket is considered of more hydrophobic in nature for being deep, less solvent exposed, and with conserved hydrophobic pocket lining residues. Being hydrophobic and with large surface area, the Mpro binding site could favour higher nonpolar interactions with N3 where the latter attained a more extended conformation within the target's pocket. Finally, similar binding pattern was depicted for Lenvatinib (4), Nintedanib (9) and 32 across the docking and MD simulation study. Compounds Lenvatinib (4) and Nintedanib (9), 28 and 32 exhibited more preferential free-binding energy as compared to N3 at Mpro binding site, the thing that confirms its superior target affinity over N3 within the preliminary docking analysis. It worth to not that the profound free-binding energy of the top-stable VEGFR-2 inhibitors correlates well with the obtained initial docking score ranking (Table 1).
| Ligand | VDWAALS | EEL | ΔG gas | ΔG solv | ΔTotal | Standard Error |
|---|---|---|---|---|---|---|
| Sorafenib | -24.1 | -13.0 | -37.1 | 20.2 | -16.9 | 0.16 |
| Sunitinib | -25.7 | -17.9 | -43.6 | 26.2 | -17.4 | 0.15 |
| Lucitanib | -31.9 | -10.0 | -41.9 | 23.7 | -18.2 | 0.09 |
| Lenvatinib | -44.8 | -20.2 | -65.0 | 33.0 | -32.0 | 0.07 |
| Axitinib | -47.3 | -15.6 | -62.9 | 33.3 | -29.6 | 0.04 |
| Apatinib | -31.3 | -12.6 | -43.9 | 27.4 | -16.5 | 0.06 |
| Cabozantinib | -34.6 | -12.3 | -46.9 | 25.6 | -21.3 | 0.07 |
| Nintedanib | -61.5 | -15.0 | -76.5 | 38.5 | -38.0 | 0.05 |
| 13 | -42.9 | -13.3 | -56.2 | 26.2 | -30.0 | 0.05 |
| 14 | -40.2 | -15.9 | -56.1 | 28.3 | -27.8 | 0.06 |
| 15 | -31.0 | -14.0 | -45.0 | 23.5 | -21.5 | 0.09 |
| 18 | -35.8 | -14.1 | -49.9 | 23.3 | -26.6 | 0.07 | 19 | -42.4 | -15.1 | -57.5 | 28.9 | -28.6 | 0.07 |
| 25 | -10.7 | -3.8 | -14.5 | 7.0 | -7.5 | 0.11 | 26 | -15.0 | -6.1 | -21.1 | 11.2 | -9.9 | 0.10 |
| 28 | -44.5 | -25.7 | -70.2 | 36.5 | -33.7 | 0.05 | 32 | -46.7 | -16.6 | -63.3 | 26.5 | -36.8 | 0.04 |
| 34 | -22.1 | -65.1 | -87.2 | 65.2 | -22.0 | 0.04 |
| N3 | -45.6 | - 23.2 | -68.8 | 37.6 | -31.2 | 0.04 |
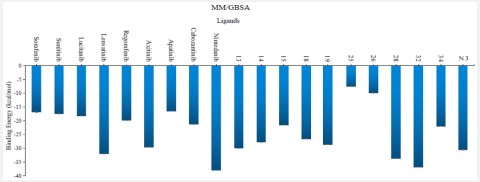
The present study suggests the potential anti-SARS-CoV-2 activities of thirty five reported VEGFR-2 inhibitors containing the amide and urea linkers. In our derivatives all thiazolidine2,4-dione derivatives are expected to be Michael acceptors and are covalent inhibitors of SARS-CoV-2. Nineteen members revealed the best in silico results [1-9, 13-15, 18, 19, 25, 26, 28, 32 and 34] and hence, were subjected to further MD simulation for their inhibitory activities against SARS-CoV-2 Mpro across the 200 ns. The molecular mechanics generalized Born surface area (MM/GBSA) calculation was implemented for binding-free energy estimation. Both compounds Lenvatinib (4), Nintedanib (9), 28 and 32 as VEGFR-2 inhibitors bound to SARS-CoV-2 Mpro target have depicted significant free binding and affinity to target's pocket. These results greatly confirmed our proposed rational and agreed with our computational findings through molecular docking (targeting the viral Mpro) and molecular dynamics which consequently suggest the strong expected activities for the other studied and discussed VEGFR-2 inhibitors as well. So, we recommend further preclinical and clinical studies for the fast repurposing of the reported, already found and discussed FDA-approved VEGFR-2 inhibitors as proposed candidates for the management of Covid-19 viral pandemic. Additionally, the studied medications can deal with as promising lead compounds for further structural modifications to enhance their activity against SARS-CoV-2.
For molecular docking studies we used AutoDock Tools software [42] and molesoft program [34]. We used the Amber 18 molecular dynamics package [43] together with the force field AMBERff14SB for the protein [44], and the GAFF2 force field for the ligands [45]. The co-crystallized inhibitor ligand (N3) was used as a reference standard.
Preparation of the tested VEGFR-2 inhibitors
Structures of the tested compounds and the formal charges on atoms were checked by 2D depiction, subjected to energy minimization and the partial charges were automatically calculated. The tested compounds together with the co-crystallized ligand (N3) were imported in the same database and saved in the form of MDB file for the docking calculations with target protease as described earlier [46].
Preparation of the target (SARS-CoV-2 Mpro)
Protein Data Bank was used to download the crystal structure of SARS-CoV-2 Mpro with (PDB code: 6LU7and resolution: 2.16 Å) [47]. The crystal structure was protonated, hydrogen atoms were added with their standard 3D geometry, automatic correction for any errors in the atom's connection and type was applied, and potential fixation of the receptor and its atoms were done. Site Finder was applied for selection of the same active site of co-crystallized inhibitor using all default items and dummy atoms of the pocket were created [48].
Docking of the tested VEGFR-2 inhibitors to the viral Mpro binding site
Docking of the previously prepared database composed of the tested thirty five VEGFR-2 inhibitors (1-35) and the co-crystallized inhibitor N3 (36) was performed. The following methodology was applied: the file of the prepared active site was loaded, and general docking process was initiated. The program specifications were adjusted so that the docking site (dummy atoms), the placement methodology (triangle matcher) and the scoring methodology (London dG). Rigid receptor as refinement methodology and GBVI/WSA dG as the scoring methodology for selection of the best ten poses from one hundred different poses for each tested compound [49]. The MDB file of the thirty ligands was loaded and general dock calculations were run automatically. The obtained poses were studied after completion, and the best ones having the best ligand–enzyme interactions and the most acceptable root mean square deviation (rmsd) values were selected and stored for energy calculations. At the beginning, a validation process was also performed for the target receptor by docking only the co-crystallized ligand and low RMSD values between docked and crystal conformations indicate a valid performance [50,51].
Molecular dynamics (MD) simulation
The top ninety ranked score complexes were processed to a molecular dynamics study to address their binding affinity to the protease enzyme. The N3 compound was also used in the docking and the simulation as a positive control. Ligand force fields were generated using GAFF2 [45] and the force field AMBERff14SB for the protein [44]. The complex was solvated expanding 15.0 Å in each direction with TIP3P water box of octahedral truncated box neutralized to a salt concentration of 150 mM of NaCl. The system was prepared via multiple energy minimization, equilibration and production steps under gradual decline position restraints on the ligand and protein achieving 310 K of temperature and 1.0 bar of pressure followed by a restraint-free production run of 200 ns. The coordinates saved every 2 ns. The binding energy between the ligand and the receptor was determined by applying MM/GBSA approach on the trajectory using MM/PBSA.py script of Amber [52], the calculations were for the last 50 ns using snapshots of 1 ns intervals from the simulation trajectory.
Conflicts of interest: The authors declare no conflict of interest.