1Department of Computer Science, City University of Hong Kong, Hong Kong, China.
2College of Chemistry and Chemical Engineering, Central South University, Changsha, China.
3College of Life Sciences, Xinyang Normal University, Xinyang, China.
Ka-Chun WONG
Email: kc.w@cityu.edu.hk
Received : Jan 18, 2023 Accepted : Feb 27, 2023 Published : Mar 06, 2023 Archived : www.meddiscoveries.org
Stomach adenocarcinoma (STAD) patients are often associated with significantly high mortality rates and poor prognoses worldwide. Among the STAD patients, competing endogenous RNAs (ceRNAs) play key roles in regulating one another at the post-transcriptional stage by competing for shared miRNAs.
In this study, we aimed at elucidating the roles of lncRNAs in the ceRNA network of STAD, uncovering the molecular biomarkers for target therapy and prognosis. Specifically, a multitude of differentially expressed lncRNAs, miRNAs, and mRNAs (i.e. 898 samples in total) were collected and processed from TCGA. Cytoplasmic lncRNAs were kept for evaluating overall survival (OS) time and constructing the ceRNA network. Differentially expressed mRNAs in the ceRNA network were also investigated for functional and pathological insights.
Interestingly, we identified one ceRNA network including 13 lncRNAs, 25 miRNAs, and 9 mRNAs. Among them, 13 RNAs were found related to the patient survival time; its individual risk score can be adopted for prognosis inference. Finally, we constructed a comprehensive ceRNA regulatory network for STAD and developed our own risk scoring system that can predict the OS time of STAD patients by taking into account the above.
Keywords: Stomach adenocarcinoma (STAD); Competing endogenous RNA (ceRNA); Prognosis; Long non-coding RNA (lncRNA); Risk model.
Stomach adenocarcinoma (STAD) is a devasting digestive tract disease that is prevalent across the world. As the fourth most frequent cause of cancer-related deaths worldwide and the fifth most common malignancy, gastric cancer(GC) continues to be a challenge for global health [20]. In 2020, there were more than 1 million new cases and an estimated 769,000 deaths, with China accounting for nearly half of both [53]. STAD makes up more than 95% of instances of GC [42]. Age, high salt intake, alcohol, and active cigarette use are all considered critical risk factors for this condition [63]. The competing endogenous RNA (ceRNA) hypothesis, an as-yet-unknown theory of how gene expression is regulated, was initially put forth by [47]. The development of STAD is strongly connected with the ceRNA regulatory network. In STAD, a prognosis-related ceRNA network has yet to be discoveredsystematically and comprehensively [40].
Non-coding RNAs having more than 200 nucleotides in length are referred to as long non-coding RNAs (lncRNAs), which lack the ability to code for proteins but have the power to control gene expression [14]. Short single-stranded RNAs called microRNAs (miRNAs), also known as non-coding RNAs, typically include 18-23 nucleotides. MiRNAs can limit the spread of cancer by acting as oncogenes or suppressors [51]. The decreased gene expression by specifically binding to the 3’UTR of downstream target mRNAs. In order to restore the activity of downstream mRNAs, lncRNAs can function as ceRNAs that bind to miRNAs [69]. Cell proliferation, migration, and invasion are some biological processes regulated by the regulatory network connecting lncRNAs, miRNAs, and mRNAs. Cell growth and development are impacted by ceRNA network disruption, which frequently results in numerous illnesses, particularly cancer [52]. In STAD, lncRNAs, miRNAs, and mRNAs work together rather than just interacting directly. For example, according to Huang et al. research, the IGF2-AS-miR-503-SHOX2 ceRNA network was responsible for IGF2-AS’s role in promoting tumor growth and invasion [18]. Zong et al. point out that the RhoA signaling pathway allowed the CTC-497E21.4-miR-22-3p-NET1 ceRNA network to play beneficial functions in the evolution of GC [71].
Researchers are increasingly adopting statistical algorithms to investigate novel diagnoses and therapy targets due to precision medicine’s quick progress. Exploring the relationship between genomic features and clinical characteristics was made possible because of the TCGA database source. The TCGA team has so far used integrated multi-dimensional analytics and wide-scale genome sequencing to examine large cohorts of more than 30 human tumors. Research on specific cancer types as well as thorough assessments of all cancers, has added to our understanding of carcinogenesis. For example, Li et al. constructed a ceRNA network using 15 differentially expressed mRNAs, one differentially expressed miRNA, and two differentially expressed lncRNAs using integrated analysis based on TCGA [34]. Liu et al. explored the interactions between differentially expressed lncRNA, miRNA, and mRNA in TCGA and established a lncRNA-miRNA-mRNA network in clear cell renal cell carcinoma [31]. Gao et al. identified early diagnostic and prognostic biomarkers for liver cancer based on studies on TCGA RNA-seq and clinical information data [13].
In this study, we systematically and comprehensively identified survival-related ceRNA networks in STAD. The study of cancer from a molecular perspective has recently been a research hotspot due to advancements in diagnostic technologies and a deeper understanding of cancer gene maps. This has also led to improvements in the study of the molecular basis of treatment for patients with STAD. A flow diagram for the Construction of the ceRNA network is shown in Figure 1.
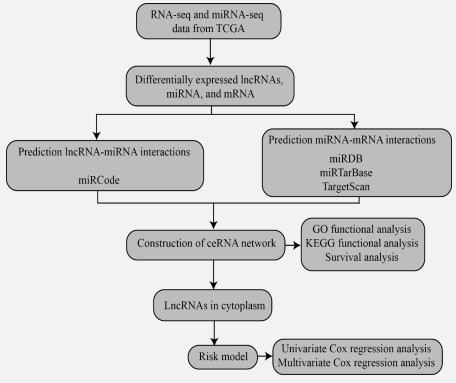
Data collection and pre-processing
We downloaded lncRNA, miRNA, mRNA expression profile, and their clinical information data from TCGA (https://www. cancer.gov/tcga) database using TCGAbiolinks (version: 2.25.0) R package [10]. For RNA-seq data (lncRNA and mRNA), we obtained 407 STAD samples (375 tumor and adjacent 32 normal samples). For miRNA-seq (miRNA), we obtained 491 STAD samples (446 tumor and 45 adjacent normal samples). The gene type information came from GENCODE (https://www.gencodegenes.org/, version: GRCh38/hg38). We defined the following six gene types as lncRNAs: sense_overlapping, lincRNA, 3prime_overlapping_ncrna, processed_transcript, antisense, and sense_intronic.
Identification of DEGs
Differentially expressed genes (DEGs) were identified with edgeR (version: 3.38.1) R package [46]. The criteria for selecting differentially expressed lncRNAs, miRNAs, and mRNAs is |log2 (fold change)|> 1 and FDR < 0.05. Three volcano plots were utilized to visualize DEGs (lncRNAs, miRNA, and mRNA). We treated the genes as up-regulated genes if they were significantly up-regulated in STAD tumor samples compared to adjacent normal samples, while we treated the genes as down-regulated genes if they were significantly down-regulated in STAD tumor samples compared to adjacent normal samples.
Interaction annotations
Interactions between lncRNAs and miRNAs: The miRCode (http://www.mircode.org/) database [21] was used for inferring putative miRNA target sites in the lncRNA. We exported the interacted miRNAs from miRCode (version: 11, June 2012) for differentially expressed lncRNAs.
Interactions between miRNAs and mRNAs: We adopted a strategy based on the vote for improving the reliability of miRNA and mRNA interactions following published methods [2,7]. If an interaction occurred in twice of the following three databases, miRDB(http://www.mirdb.org/, version: 6.0, June 2019) database [9], miRTar Base database (https://mirtarbase.cuhk. edu.cn/~miRTarBase/miRTarBase_2022/php/index.php, version: 9.0, September 2021) [17], and TargetScan (https://www. targetscan.org/vert_80/, version: 8.0, 23 May 2022) database [37], we picked up this interaction.
Subcellular localization of lncRNAs
We used the lncLocator (http://www.csbio.sjtu.edu.cn/bioinf/lncLocator/) database [5] to look into the lncRNAs' intracellular location because they can only serve as nodes of the ceRNA network in the cytoplasm. The locLocator is an ensemble classifier-based predictor exploiting lncRNA sequence information. So, we exported the sequences from the GENCODE database's lncRNA transcript sequence information (version: GRCh38/ hg38). Then, lncRNA sequences were uploaded into the lncLocator database. The subcellular locations consist of cytoplasm, nucleus, cytosol, ribosome, andexosome in the locLocator.
Construction of ceRNA network
Cytoscape (http://www.cytoscape.org/, version 3.9.0) [48] software was utilized for visualizing the locations of lncRNAs and ceRNA networks.
Functional enrichment annotation of DEGs
We used DAVID (The Database for Annotation, Visualization, and Integrated Discovery, https://david.ncifcrf.gov/) to carry out GO enrichment analysis in order to investigate the role of differentially expressed mRNAs in the ceRNA network [16,50]. The chord diagrams were created using the GOplot R package (version 1.0.2). In addition, the KEGG (Kyoto Encyclopedia of Genes and Genomes, https://www.genome.jp/kegg/) signaling pathway enrichment analysis of mRNAs in the ceRNA network was carried out using the cluster Profile (version 4.4.4) package in R [41,24,22,23,25].
Optimal cutoff selection and Kaplan-Meier survival analysis
There is no difference in survival analysis in many cases when using specific locations, such as median, mean, quartile, etc., as the cutoff point. At this time, it is often necessary to find an optimal cutoff point to make a difference in survival analysis. The surv_cutpoint() function in survminer (version: 0.4.9) package R was utilized to display the distribution of gene expression levels and determine the optimal cutoff for survival analysis [30,54]. R package survival (version: 3.3.1) was used for visualizing the difference between low- and high-expression groups classified by the obtained optimal cutoff. P-values below 0.05 were regarded as significant.
Creation of risk scoring model
The upstream region of the ceRNA network is dominated by lncRNAs, which serve as the main miRNA and mRNA effectors [67,28]. Additionally, lncRNAs are very particular in their expression and distribution, making them the best possible biomarkers for diagnosing and evaluating the prognosis of STAD [6,15]. The survival (version: 3.3.1) R package was used to create a risk-scoring model for lncRNAs in the ceRNA network.The multivariate Cox regression model included LINC00486 and LSAMP-AS1 to make the independent prognostic signature for STAD (P-value ≤ 0.05). The following formula was used to calculate the risk score for each patient:
risk score =
Here, represents the expressionlevels of lncRNA. β represents the regression coefficient of multivariate Cox regression
for lncRNA.
Univariate and multivariate Cox regression Mining the independent factors is essential for prognostic analysis for cancer research [32]. Univariate Cox regression analysis was conducted to determine whether the clinical characteristics, such as age, gender, stage, tissue of origin, primary diagnosis, AJCC T, AJCC M, AJCC N, race, and risk score, were significantly associated with overall survival (OS) in STAD patients (P-value ≤ 0.05). Then, all clinical factors were compared at one time to identify independent prognostic factors (P-value ≤ 0.05). The coxph() function in survival (version: 3.3.1) R package was applied for univariate and multivariate Cox regression analysis. And forestplot(version: 2.0.1) R package was applied for visualizing the results.
Linear regression analysis of lncRNAs and mRNAs
The ceRNA mechanism theory states that lncRNAs interact directly with miRNAs to influence mRNA expression favourably [65]. Using R software and ggpubr (version: 0.4.0) package, linear regression analysis of the log2 transformed normalized the ceRNA network's lncRNA, and mRNA expression levels were performed. The results were displayed via ggscatter() function. We regarded there as a statistically significant correlation between lncRNA and mRNA if their P-value ≤ 0.05 and cor> 0.3.
Differentially expression of lncRNA, miRNA, and mRNA
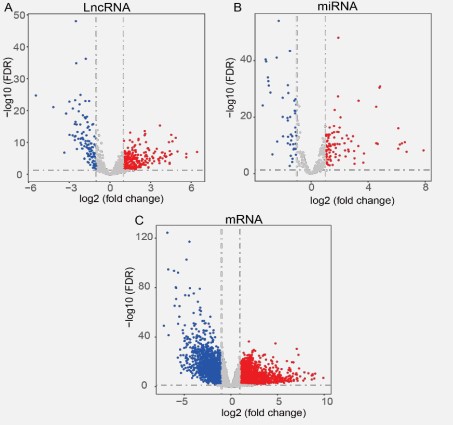
One can study the differences between tumor and adjacent normal samples to determine the genetic origin and biological pathways, therefore, identifying potential targets for treating cancer [39]. Identification of DEGs is vital for cancer research [66]. From the TCGA database, we obtained the expression profiles of 2,366 lncRNAs, 1,881 miRNAs, and 19,431 mRNAs. With the criteria of |log2 (fold change)|> 1 and FDR < 0.05, a total of 380 differentially expressed lncRNAs, 143 differentially expressed miRNAs, and 4,344differentially expressed mRNAs were selected.
Two hundred and seventy differentially expressed lncRNAs, 95 differentially expressed miRNAs, and 2,220 differentially expressed mRNAs were among those that were up-regulated in STAD tumor samples compared to adjacent normal samples, and 110 differentially expressed lncRNAs, 48 differentially expressed miRNAs, and 2,124 differentially expressed mRNAs were among those that were down-regulated (Figure 2A-C). They were regarded as essential genes involved in the early incidence of STAD.
Interactions between lncRNA and miRNA
Using the miRCode database, we firstly identified possible miRNAs interacting with 380 DE lncRNAs. The interacting genes between the 143 DE miRNAs and the predicted miRNAs were then identified. Finally, we discovered 28miRNAs and 26lncRNAs with the potential for mutual interaction (Table S1).
Interactions between mRNA and miRNA
By using mRNAs targeted by miRNAs shared by two out of three databases (miRDB, miRTarBase, and TargetScan), we identified target genes for the 26 miRNAs mentioned above, which increased the accuracy of the bioinformatics prediction. Then, we contrasted potential target mRNAs with 1,535 mRNAs that had differential expressions. Finally, the ceRNA network was established through miRNA-mRNA interaction pairs, including 16 mRNAs and three miRNAs (Table S2).
Subcellular localization of lncRNA and construction of ceRNA network
The endogenous competition role of lncRNAs is mainly manifested in the cytoplasm, making it imperative to examine the cytoplasmic-nuclear localization of these lncRNAs in order to understand their complex but precise regulatory processes better [12]. We counted and drew the distribution of 26 lncRNAs using Cytoscape.In this study, we identified 13 lncRNAs located in the cytoplasm of the cell using lncLocator software (Figure 3A and Table S3).
After taking into account the interactions between the remaining DEGs, a final STAD ceRNA regulatory network with 47 nodes and 98 edges was created by combining 13lncRNAs, 25 miRNAs, and nine mRNAs. The ceRNA network was visualized by Cytoscape software (Figure 3B).
GO term and KEGG pathway
The nine mRNAs in the ceRNA regulatory network were investigated concerning their putative biological functions and pathways. We conducted GO functional enrichment analysis on the DAVID database and reported significant enriched GO terms. There are 18 significant GO terms enriched by mRNAs in the ceRNA network (P-value ≤ 0.05, Table S4). We used a chord figure to visualize the enrichment results (Figure 3C). The top five GO terms among these were "transcription factor activity, sequence-specific binding", "nucleoplasm", "chromatin", "RNA polymerase II core promoter proximal region sequence-specific DNA binding", and "positive regulation of cell proliferation".
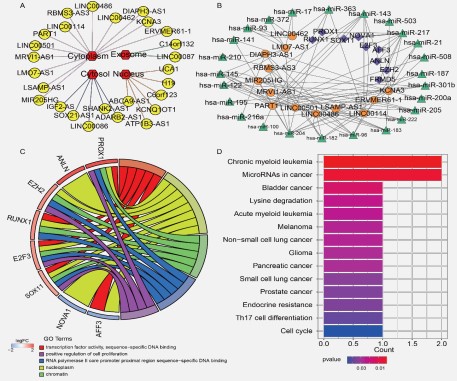
KEGG was also selected for annotation of the function of mRNAs in the ceRNA network. The R package clusterProfiler was utilized to perform gene enrichment analysis and visualize the results (Figure 3D). mRNAs in the ceRNA network enriched in 14 significant KEGG signaling pathways (P-value ≤ 0.05). The top five KEGG pathways among these were "Chronic myeloid leukemia", "MicroRNAs in cancer", "Bladder cancer", "Lysine degradation", and "Acute myeloid leukemia".
Survival curves of ceRNA network-related genes
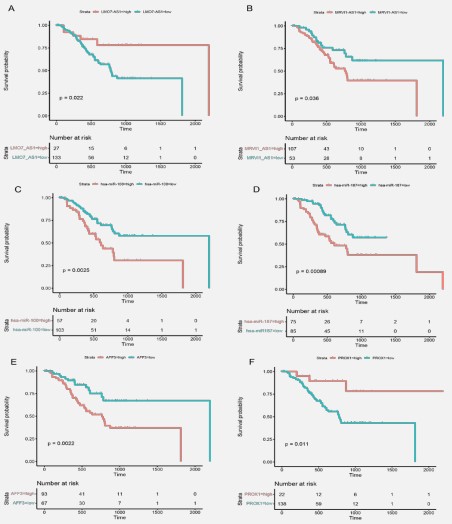
Kaplan-Meier survival analyses and log-rank tests for each gene were carried out to assess the contributions of gene expression levels to global OS time [32]. Thirteen lncRNAs, 25 miRNAs, and nine mRNAs were put into this survival analysis model in order to identify the probable genes with substantial associations with the prognostic characteristics of patients with STAD. The cutoff value was set based onsurv_cutpoint() function from survminer R package. There are 18 genes, including twolncRNAs, 12 miRNAs, and four mRNAs,that significantly differ between low- and high-expression levels groups (P-value ≤0.05, Table S5). We selected two genes for each gene type (lncRNA, Figure 4(A-B): LMO7-AS1 and MRVI1-AS1; miRNA, Figure 4C-D:hsa-miR-100 and hsa-miR-187; and mRNA, Figure 4E-F: AFF3 and PROX1) to illustrate the difference in survival probability grouped by gene expression levels.
Construction of risk score model
LncRNAs, which act as the primary miRNA and mRNA effectors, predominate in the upstream area of the ceRNA network [67,28]. Also, lncRNA hashighly specific expression distribution patterns, making them the ideal biomarkers for identifying STAD and guiding its prognosis [6,15]. Therefore, multivariate Cox regression analysis was used to determine possible prognostic-related lncRNAsbased on the 13 lncRNAs in the ceRNA regulatory network, and their contributions were weighted by their relative coefficients.The risk score model was then updated to include LINC00486 (P-value = 0.031 and HR = 1.002 (1.000-1.003)) and LSAMP-AS1 (P-value = 0.047 andHR = 1.008 (1.000-1.015)) and the final risk scoreformula was as follows:
risk score =
1.61 × 10−3 × 𝑡ℎ𝑒 𝑒𝑥𝑝𝑟𝑒𝑠𝑠𝑖𝑜𝑛 𝑙𝑒𝑣𝑒𝑙𝑠 𝑜𝑓 𝐿𝐼𝑁𝐶00486 +
7.67 × 10−3 × 𝑡ℎ𝑒 𝑒𝑥𝑝𝑟𝑒𝑠𝑠𝑖𝑜𝑛 𝑙𝑒𝑣𝑒𝑙𝑠 𝑜𝑓 𝐿𝑆𝐴𝑀𝑃 − 𝐴𝑆1
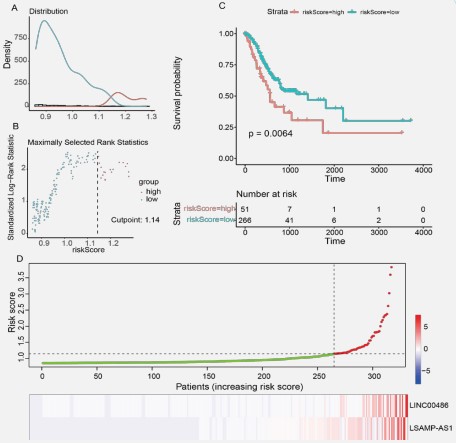
Positive coefficients were found for LINC00486 and LSAMPAS1 in both the univariate and multivariate Cox regression analyses. This phenomenon suggested that they are cancer risk factors for the survival time in STAD.As shown in Figure5(A-B), the distribution of all risk scores and maximally selected rank statistics were analyzed by surv_cutpoint() function in the survival R package. Thus, we set 1.14 as the optimal cutoff. Patients with risk scoresless than or equal to the cutoff were assigned to the low-risk group (315 patients), and those with risk scores greater than the cutoff were categorized into the high-risk group (60 patients), respectively. The risk score signature's Kaplan-Meier survival analysis revealed a significant difference in survival times between the low- and high-risk score groups (P-value ≤ 0.05, Figure 5C). Scatter plot and heatmap showed the lncRNAs' expression profiles and risk scores of 315 patients with survival time (Figure 5D). The results revealed that patients' risk scores increased as the expression levels of lncRNAs.
Identification of independent prognostic factors
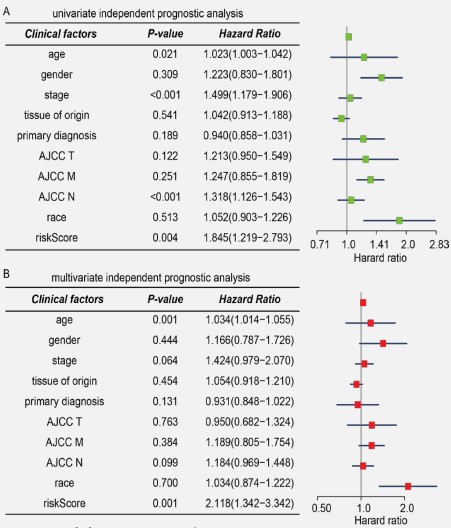
Following this, a univariate Cox regression analysis was carried out to examine the 317 patients with complete clinical data for any markers that might be associated with OS. Based on the following clinical traits, we separated the patients into different groups: age, gender, stage, tissue of origin, primary diagnosis, AJCC T, AJCC M, AJCC N, race, and risk score. The finding demonstrated that, like risk score, the age, stage, and AJCC N had statistically significant prognostic values in the green forest plot (Figure 5A).
The red forest plot demonstrated that stage and AJCC N clinical factors were not linked to the prognosis of STAD patients in the multivariate Cox regression analysis. Thus, an independent predictive indicator of survival time for STAD patients was the risk score system created from the expression levels oflncRNAs in the ceRNA regulatory network (Figure 5B).
Relationships between lncRNA and mRNA
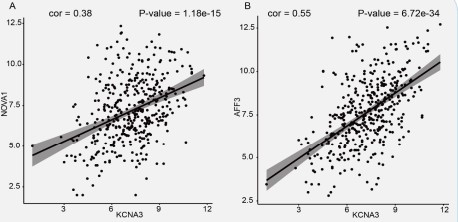
Under the ceRNA mechanism concept, lncRNAs interact directly with miRNAs to positively affect mRNA expression [65]. Regression analysis of 13 lncRNAs and 9 mRNAs was performed to confirm this positive relationship phenomenon in STAD. The correlation values and P-values were calculated and counted. There are 21 positive correlation pairs, including eight lncRNAs and nine mRNAs (P-value ≤ 0.05 and cor> 0.3, Table S6). Furthermore, we explored to see if the lncRNAs and mRNAs shared any miRNAs. The findings demonstrated the importance of hsamiR-217 as a critical miRNA in several ceRNA pathways, including KCNA3-hsa-miR-217-AFF3 and KCNA3-hsa-miR-217-NOVA1. We plotted the linear regression curves between KCNA3 and mRNAs in Figure 7(A-B).
As the fourth most frequent cause of cancer-related deaths worldwide and the fifth most common malignancy, STAD continues to be a challenge for global health [20]. Patients with STAD continue to have a poor prognosis and an inadequate survival rate despite large improvements in diagnosis, prevention, and therapy [61]. The ceRNA regulatory network is crucial to cancer development [52]. ceRNA network disruption affects cell growth and development, leadingtovarious diseases, including cancer [52]. Unfortunately,very littleresearch has been performed on the ceRNA regulatory network in STAD prognosis.
Interestingly, recent research has provided fresh insight into the role of lncRNAs in the development of STAD [35]. In the tissues of STAD, a sizable number of miRNAs exhibit variable expression (Wu et al. 2010). Protein-coding proto-oncogenes and tumor-suppressor genes are altered genetically and epigenetically during the multi-step process of STAD [19]. In particular, cytoplasmic lncRNAs are essential for a variety of molecular processes in both animal and human cells. They have the ability to influence mRNA stability, control mRNA translation, act as ceRNAs, act as miRNA precursors, and mediate protein changes [45]. Certain cytoplasm lncRNAs can control how cytoplasmic proteins are transported into the nucleus to activate transcription [58]. Thus, we focus on the lncRNA-miRNA-mRNA regulatory ceRNA network in this research. Also, we find an optimal cutoff point to make a difference in survival analysis. In which we only selected lncRNA,whichis located in the cytoplasm.Based on survival analysis, LMO7-AS1 and MRVI1-AS1 were recognized as possible prognostic biomarkers and therapeutic targets in STAD. High expression of LMO7-AS1 was linked to a poor prognosis in patients with childhood kidney cancer, according to research by Zheng et al. [68]. LMO7-AS1 was up-regulated in colorectal cancer (CRC) tumors compared to non-tumor tissues [60]. Qin et al. reported that LMO7-AS1 was positively correlated with poor prognosis of CRC by screening 240 differentially expressed immune-associatedlncRNAs between CRC tissues and normal tissues using the bioinformatics method [44]. The survival analysis identified has-miR-100 and has-miR-187 as potential prognostic biomarkers in STAD. miR-100 is the most important miRNA related to the progression of GC [57]. MD et al. suggested that by performing cluster analysis, miR-100 is up-regulated in diffusetype GC [57]. miRNA-100 was also up-regulated in pancreatic Adenocarcinoma [4]. By blocking FOXA2, miR-187 encourages the growth and spread of GC [27]. In GC, miR-187 may serve as a biomarker and therapeutic target [8]. AFF3 and PROX1 have been deemed to be candidate prognostic biomarkers in STAD on the basis of survival analysis. In a recent study, AF4/FMR2 family member 3 (AFF3) was proposed by Zeng et al. as a novel prognosis-related biomarker that can be used for immunotherapyby an integrated multi-omic framework [64]. AFF3 has recently been discovered to play a significant role in the initiation and growth of several malignancies, including glioma, breast cancer, and adrenocortical carcinoma [64].
Additionally, the Cox regression analysis got the lncRNAs LINC00486 and LSAMP-AS1 from the study of 13 lncRNAs. MicroRNA-182-5p may be a target gene for LINC00486, according to bioinformatics analysis, and gastric cancer cells have higher levels of this RNA, which can encourage cancer cells' invasion and migration [33]. Ma et al. concluded a novel lncRNA LSAMP-AS1 is involved in the prostate cancer process via targeting miR-183-5p/DCN axis, and they reported thatLSAMP-AS1 binds to microRNA-183-5p to suppress the progression of prostate cancer by up-regulating the tumor suppressor DCN [36]. The ceRNA regulatory network's dysregulated mRNA function is mainly located in the following GOterm:transcription factor activity, nucleoplasm, chromatin, RNA polymerase II core promoter proximal region sequence-specific DNA binding, and positive regulation of cell proliferation. PROX1, AFF3, SOX11, E2F3, and RUNX1 were involved in the transcription factor activity GO term. One of the most important controls over mammalian expression is runt-related transcription factor 1 (RUNX1) [56]. In cohorts of glioma, pancreatic cancer, colorectal cancer, cervical cancer, renal cancer, lung cancer, ovarian cancer, and gastric cancer, RUNX1 is a worse predictive indicator [29]. On the other hand, better clinical outcomes are associated with patients with higher levels of RUNX1 expression in both breast and prostate cancer patients [29]. By targeting RUNX1 and activating the Hippo signaling pathway, miR-28b-3p prevents invasion, migration, and epithelial-mesenchymal transition in GC [3]. SOX11, E2F3, EXH2, and PROX1 were involved in the positive regulation of cell proliferation GO term.Due to its methyltransferase activity, Polycomb group protein enhancer of zeste homolog 2(EZH2), a member of the PRCs family, can play functional roles in the cell. By stimulating H3K27me3, EZH2 influences gene expression(Mirzaei et al. 2022).EZH2 inhibitors have reportedly reduced metastasis and neovascularization in human malignancies. EZH2 inhibitors have been used to treat colon cancer because they cause tumor cells to respond to them more frequently (stage II and III) [38].
In particular, we have identified two promising ceRNA networks: KCNA3-has-miR-217-NOVA1 and KCNA3-has-miR-217-AFF3, involved in STAD progression and prognosis.Also known to be dysregulated in PCa are the following K+ channels, which have been suggested as biomarkers. In high-grade malignancies, Potassium Voltage-Gated Channel Subfamily A Member 3(KCNA3) is down-regulated and mostly expressed in the early stages of cancer growth [1]. LncRNA KCNA3 serves as a potential prognostic biomarker and therapeutic target for CRC since it inhibits the growth of tumors by downregulating the expression of YAP1 [70]. When compared to the normal stomach gland tissues, KCNA3 has been found and is significantly downregulated in stage-I STADs, which suggests it is very useful as an early diagnostic indicator [55]. In cancer, miR-217 is typically dysregulated. Chen et al. demonstrated that tumor tissue has lower levels of miR-217 than the nearby normal tissue [11]. In patients with GC, a low expression level of miR-217 was linked to aggressive tumor characteristics and poor OS [11]. The expression of neuro-oncological ventral antigen 1 (NOVA1) was suppressed in GC, and ectopic NOVA1 expression in tumor cells contributes to tumor growth and a poor prognosis [26]. Reduced NOVA1, which was suppressed by miR-146b-5p, is a possible biomarker for predicting poor prognosis in individuals with GC [62]. It is also a biomarker of concealed residual disease in leftover tissues following GCresection [62]. Furthermore, Shen et al. showed that downregulating NOVA1 by miR-339 overexpression in GC cells inhibits malignant characteristics like proliferation, invasion, migration, and oncogenicity while restoring NOVA1 in miR-339-overexpressing GC cells partially reverses the inhibitory effects of miR-339 [49].
Abbreviations
TCGA: The Cancer Genome Atlas; STAD: Stomach Adenocarcinoma; GC: Gastric Cancer;
LncRNA: Long Non-Coding RNA; miRNA: microRNA; ceRNA: Competing Endogenous RNA; GO: Gene Ontology; FDR: False Discovery Rate; KEGG: Kyoto Encyclopedia of Genes and Genomes; DEG: Differentially Expressed Gene; DAVID: The Database for Annotation, Visualization and Integrated Discovery; AJCC: The American Joint Committee on Cancer; CRC: Colorectal Cancer; OS: Overall Survival.
Author contributions: KW conceived and performed the study conception and design. ZL performed data collection and analysis. ZL and FL wrote the first draft of the manuscript. OP, YZ,and HW were involved in the grammar-checking part. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Institutional review board statement: Not applied.
Data availability statement: The public availability dataset of this research is from the accessibility database. The lncRNA, mRNA, miRNA expression profiles, and clinical data were downloaded from the TCGA Research Network: https://www.cancer. gov/tcga.
Funding: This study received considerable support from the Shenzhen Research Institute, City University of Hong Kong, as well as funding from two research programs granted by the National Natural Science Foundation of China (Grant Nos. 32170654 and 32000464). A grant from the Food and Health Bureau, the Health and Medical Research Fund, and the Government of the Hong Kong Special Administrative Region [07181426] provided significant funding for the activities reported in this publication. The work described in this paper was partially supported by grants from the City University of Hong Kong (CityU 11202219, CityU 11203520, CityU 11203221).
Declaration of competing interest: The authors have no relevant financial ties to or affiliations with any company or organization that would have a financial stake in or a conflict of interest with the materials included in the article.
Acknowledgments: RNA-seq data and the associated clinical data of the STAD patients were generated by the TCGA Research Network and are available at https://www.cancer.gov/tcga. We appreciate the technical assistance from Shengxin.Ren firm and the server computing resource help from the Tongyuan gene company.
Appendix supplementary materials
Supplementary Table 1. The interactions between lncRNAs and miRNAs.
Supplementary table 2. The interactions between miRNA and mRNAs.
Supplementary table 3. Intracellular localization of lncRNAs.
Supplementary table 4. GO functional enrichment analysis results.
Supplementary table 5. Survival analysis of ceRNAs network-related genes.
Supplementary table 6. Correlations of lncRNA-mRNA pairs.