Department of Integrative Biomedical Sciences, Faculty of Health Sciences, University of Cape Town, South Africa.
Chacha M Issarow
Email: chacha.issarow@uct.ac.za
Received : Feb 12, 2026 Accepted : Mar 09, 2026 Published : Mar 16, 2026 Archived : www.meddiscoveries.org
Background: Whole-Genome Sequencing (WGS) is a powerful tool in the investigation of Tuberculosis (TB) transmission and disease recurrence due to its high discriminatory power compared to other genotyping methods. This study aimed to review methods used to assess and describe the transmission dynamics of Mycobacterium Tuberculosis (MTB) in low and high TB incidence settings using WGS data.
Method: Four electronic databases were used for searching studies that used WGS to assess TB transmission either in low or high TB burden settings. Studies published in English from 2008 onward applying WGS to describe TB transmission dynamics in humans were eligible for review.
Results: The majority of studies reviewed (28 studies) used single nucleotide polymorphism (SNP) threshold methods to assess and describe TB transmission by computing genetic distance between paired isolates, based on the notion that shorter genetic distances indicate recent TB transmission. Of 28 studies that used the SNP threshold approach, eight studies also used the presence of drug resistance conferring mutations to assess and describe recent TB transmission, assuming that isolates with the same mutations originated from the same source. Three studies developed new methods to assess and describe TB transmission, of which two studies formed transmission trees using novel Bayesian and epidemiological model approaches, and one study identified clusters based on probabilistic matching. One study assessed and described TB transmission based on SNP distances by determining the number of isolates per isolate in a range of ≤10 SNPs referred to as transmission index.
Conclusion: While all methods have technical pros and cons, the SNP threshold method was primarily used to investigate TB transmission in both low and high TB incidence settings. Contact tracing was applied in very few studies based in low incidence settings as it is challenging in high incidence areas due to the overall TB burden.
Keywords: Mutation; Recent TB Transmission; Drug Resistance; SNP Threshold.
Tuberculosis (TB) remains a global public health concern, particularly in low- and middle-income countries [34]. The emergence of Drug-Resistant TB (DR-TB), including Multidrug Resistant (MDR) and extensively Drug-Resistant TB (XDR-TB) remains a challenge in TB treatment and control worldwide [34,46,47]. Due to the complex nature of TB, including DR-TB, traditional epidemiological methods are insufficient to infer the exact transmission in populations. The complexity of the TB epidemic is due to the fact that the majority of exposed or infected individuals do not develop disease, and that there is often a long-time frame between exposure and disease. Additionally, TB complexity may be influenced by many other factors, including the biology of the bacillus, failure of the public health care and comorbidities, such as HIV. Reducing ongoing transmission is key to effective control of TB [60]. A better understanding of how, where and when TB is transmitted would help to triage appropriate strategies for prevention of transmission.
Previously, it was assumed that DR-TB was mainly attributable to the acquisition of drug resistant conferring mutations during inadequate therapy or poor treatment adherence [47]. However, recent modelling and epidemiological data show that drug resistance caused by direct transmission of already drug-resistant TB strains from person-to-person plays an important role even among previously treated cases [4,20,22,38,45]. Additionally, molecular epidemiological studies have demonstrated that direct transmission can account for up to 84% of the notified DR-TB cases, particularly in high burden regions [51]. Mycobacterium Tuberculosis (MTB) comprises different phylogenetic lineages [39], and it was suggested that genetic differences between bacterial clades could account for different epidemiological characteristics such as transmission [36,37,39]. Additionally, different evolutionary rates of MTB strains might be among the key factors influencing the genetic diversity of these bacteria [31,62].
Due to decreasing costs, Whole-Genome Sequencing (WGS) is becoming more widely applied to study TB transmission as well as the evolution of drug resistance, and has begun to inform knowledge around these important aspects of TB pathogenesis [7,19]. WGS identifies sequence variations at the whole genome level, and, therefore, has the potential to help in quantifying and describing transmission with more accuracy compared to traditional genotyping methods [7,19,22,56]. Compared to other existing genotyping methods, WGS is better able to discriminate between relapse and re-infection mechanisms of TB recurrence, especially when patients are re-infected with closely related strains [40,41,58], identify transmission chains [2,21], measure within-host diversity [63-67], and differentiate between direct transmission and acquired drug resistance [19,52]. WGS is therefore increasingly being used to predict epidemiological links between TB cases and to assist in transmission investigation and interruption [43,48,49,54,57 59]. Researchers have used a range of methods to assess TB transmission across low and high TB burden settings and with respect to TB drug resistance. This review aimed to describe all the methods that used WGS to assess TB transmission (drug susceptible or DR-TB) either in low or high TB burden settings, and to describe potential challenges and limitations.
Search strategy: Four electronic databases (PubMed, Scopus, Escudos and Web of Science) were searched for cross-sectional and cohort studies published from 2008 onward, assessing and describing TB transmission using WGS. In the database, the keywords “Tuberculosis”, or “TB”, and “transmission”, and “whole genome sequencing” were used for searching.
Review criteria: All articles published in English describing TB transmission in humans using WGS data either qualitatively or quantitatively were eligible for review. Therefore, publications aligned with our review criteria of assessing or describing TB transmission dynamics using WGS published in English from 2008 onward were identified and imported into Zotero for references. Review and articles with no primary data were excluded.
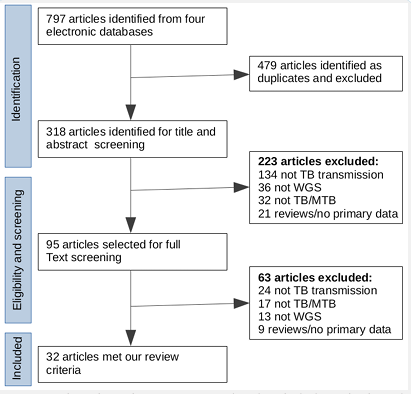
Study identification and review: Based on our review search key terms, a total of 798 publications were identified in the four electronic databases. Of the 798 identified articles, 480 were excluded as duplicates, and 318 articles remained for title and abstract screening. Of the 318 articles screened, 95 articles were selected for full text screening, of which 32 met our review criteria (Figure 1). Of the 32 studies included, 17 referred to DR TB and the remaining 15 studies referred to Drug Susceptible TB (DS-TB). Of the 32 studies that met our review criteria as mentioned in the methods, 14 studies were conducted in low (high-income countries) and 18 studies in high (low-income countries) TB burden settings [16,34].
Of the 32 studies reviewed in this study, genetic distance between paired isolates (SNP threshold) and similarity of drug resistance conferring mutations were among the most common methods used to infer recent or direct transmission of TB strains from person-to-person [20]. A total of 28 studies applied SNP threshold method, of which eight studies also used presence of drug resistance conferring mutations to confirm recent or direct TB transmission, and two studies [27,28], used both SNP threshold and newly developed methods to assess and describe TB transmission. Four [29-32], of 32 studies developed new methods to assess and describe TB transmission.
TB transmission description using the SNP threshold approach: The SNP threshold (defined as a cut-off number of SNPs that differ between paired isolates) places two or more individuals in the same transmission cluster if there is a shorter genetic distance (number of SNPs that differ between two sequences) than a specified SNP threshold between their genomes. One study [28], assumed that paired isolates or cluster of gnomically linked cases with a genetic distance of < 6 SNPs confirm recent TB transmission. Clustering isolates with shorter genetic distances than a specified SNP threshold together with identical drug resistance conferring mutations was used in some studies to define recent TB transmission. For example, one study [22], found that 32% of MDR strains were in a cluster that differed by ≤12 SNPs, indicating recent transmission of MDR strains. However, in some clustering methods, a pair of isolates with a greater genetic distance than a specified SNP threshold might be in the same transmission cluster if linked by chains of intermediate unsampled cases [31], which highlights that assessment of transmission is highly dependent on the completeness of sampling.
Of 32 studies reviewed in this study, the majority (28 studies) used the SNP threshold method to assess and describe TB transmission [24]. Specifically, five studies [3,9-12,21], used a genetic distance of ≤3 SNPs, ten studies [1-11,14,15,19,26], used a genetic distance of ≤5 SNPs, four studies [1,6,16,27], used a genetic distance of ≤10 SNPs to confirm recent or direct transmission and four studies [13,17,20,22], used a genetic distance of ≤12 SNPs to form transmission clusters. Of these four studies that used ≤12 SNPs to infer transmission, three studies [13,17,20], combined contact tracing and WGS to confirm recent TB transmission, and were conducted in a low-TB burden setting. Contact tracing complemented with genotyping of MTB isolates is important for understanding disease transmission [17]. The SNP threshold method was therefore used together with contact tracing in three reviewed studies from low incidence settings to assess and describe TB transmission. However, TB contact tracing is mainly used in low incidence settings (high-income countries), as it is challenging in high incidence areas due to the overall TB burden [15]. However, TB contact screening is primarily household based in high incidence settings and often focusses on children.
Bjorn-Mortensen et al [9], suggest that defining a minimum distance of 12 SNPs as a threshold for unlikely recent transmission and a maximum distance of 5 SNPs as the threshold for likely transmission is adequate in low incidence settings, but such a threshold is more difficult to define in high TB burden settings. In high burden settings, certain strains may have been circulating for a long time, and thus the MTB diversity will be limited with many of these “endemic” strains differing by fewer than 12 SNPs. Many of the studies reviewed here used WGS based on the SNP threshold approach to suggest TB transmission without comparison to epidemiological contact data. However, differences in sample processing prior to WGS, variability in data analysis approaches (pipelines) and differences in sampling intensity remain significant challenges [31,42]. Multiple sample processing and WGS data analysis pipelines exist that differ widely in output formats, making SNP threshold standardization difficult even in the same setting [42].
TB transmission assessment using drug resistance conferring mutations: Apart from the SNP threshold approach, comparison of first-line or second-line drug resistance conferring mutations was used in eight studies to infer recent or direct TB transmission [8]. It is assumed that isolates with the same drug resistance mutational profile originated from the same common ancestor (if only DR strains are sampled and the SNP threshold is below a certain value), which indicates recent or direct transmission [8]. Of the 32 studies reviewed, eight studies [19-26], used clustering based on both SNP thresholds and comparison of drug resistance conferring mutations to assess TB transmission. For example, using a genetic distance of ≤12 SNPs and assuming that resistance via canonical mutation does not occur in parallel, Yang et al [22], found that 89.5% of the genomic clusters had resistance mutations for isoniazid and rifampicin that were consistent among clustered strains, confirming direct transmission of MDR strains rather than acquired resistance. Additionally, Caselli et al [21], found that strains containing the same mutations conferring resistance to rifampicin, isoniazid, streptomycin and ethambutol clustered phylogenetically, thus confirming direct transmission of DR TB strains from an inferred common ancestor. Using a genetic distance of <50 SNPs to define transmission clusters, Clark et al [23], found that isolates in the same cluster had almost (not all) identical mutations conferring resistance to rifampicin and isoniazid, suggesting direct transmission of MDR-TB. However, this study (Clark et al) did not require that all isolates within a cluster had identical drug resistance conferring mutations to confirm transmission of MDR strain. This implies the fact that there are highly common drug resistance conferring mutations in clinical isolates, so that having the same mutations in clustering isolates does not necessarily mean that transmission took place.
The majority of reviewed studies used the MTB H37Rv, which is a lineage 4 strain [42], as the reference genome to identify site specific sample-specific genomic variations. However, one of the reviewed studies [18], suggested that existing methods for comparative analysis of isolates of using a single MTB strain, such as H37Rv, as the reference genome, may limit resolution (discriminatory power). The study [18], therefore used a pan genome reference and formed clusters based on SNP threshold approach to assess TB transmission and compared results with other methods that used the MTB H37Rv strain as the reference. The pan-genome is derived from more than 100 MTB reference genomes representing lineages 1-4 for read mapping prior to variant calling. This approach allows the comparison and clustering of a large number of diverse samples using a pan-genome reference sequence inferred computationally. Using a genetic distance of <13 SNPs for transmission cluster link detection, the authors suggested that the pan-genome approach is superior to previously published methods in several datasets and across different MTB lineages, as its characteristics allow the comparison of a high number of diverse samples in one analysis [18].
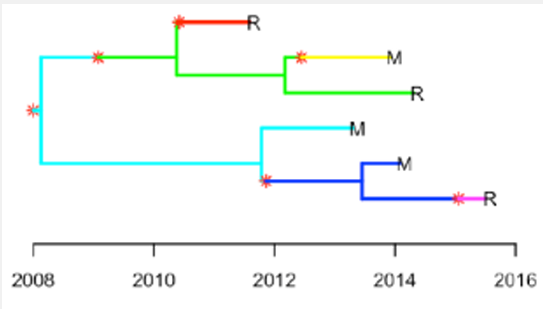
TB transmission assessment using newly developed methods: Diderot and colleagues [29], developed a new Bayesian based method for inferring TB transmission called Transpyloric. The method uses a Susceptible, Infected, removed (SIR) epidemiological model assuming that the transmission bottleneck is complete (only a single pathogen variant is transmitted from infector to susceptible per transmission event), and that all cases comprising an outbreak have been sampled. In their approach, they constructed a timed phylogenetic tree using Bayesian evolutionary analysis by sampling trees (using BEAST software), which becomes an input for Transpyloric to infer transmission networks via Markov Chain Monte Carlo simulation. The robustness of the method is based on the fact that it takes into consideration within-host diversity, which has been shown to be significant for MTB in some settings [65]. The main difference from a typical BEAST output is that Transpyloric infers a transmission tree that defines specific transmission events or new infections indicated by red stars and a change in branch colour (Figure 2). The method also produces a transmission tree from the original phylogeny with vertical arrows representing the occurrence of transmission from person-to-person [27,33]. The difference between transmission tree and phylogenetic tree is that a transmission tree can be inferred from a phylogeny while accounting for within-host genetic diversity by colouring the branches of a phylogeny according to which host those branches were in [29].
Some studies used SNP threshold combined with other new methods, such as phylogenetic modelling and a Transpyloric approach to assess and confirm recent TB transmission. For example, Yang et al [27], used both SNP threshold (≤10 SNPs) and Transpyloric methods to assess TB transmission dynamics among internal migrants in Shanghai, China. The study quantified the relative importance of latent TB importation with that of local transmission by comparing the approximated time of transmission (using Transpyloric) with reported time of arrival in Shanghai. Based on the combination of these methods (SNP threshold and Transpyloric), the authors found that the primary mechanism driving local incidence of TB in Shanghai was locally transmitted between both migrants and residents [28]. Additionally, Arabian et al [28], used SNP threshold (< 6 SNPs and 6 - 12 SNPs) and Transpyloric methods to assess TB transmission among immigrants and local individuals in Norway. The study estimated that about 25% of the patients contracted TB (via direct transmission) after having lived in Norway for almost 20 years.
Eldholm et al [30], developed a new method using epidemiological modelling approach to explore the transmission of MDR-TB and HIV co-infection. The overall aim of the study was to apply a newly developed method to explore the impact of HIV on TB transmission, MTB evolution and whether HIV co infection accelerates drug resistance evolution. The authors considered a SEIR (susceptible, exposed, infected, removed) epidemiological model and assumed that within-host diversity happens at a constant rate, α as applied in Transpyloric method [29]. The SEIR epidemiological model assumed in the development of the method implies that there is random mixing between the individuals, with every infectious individual being equally likely to infect any susceptible individual.
The Eldholm et al [30], method described above is an extension of the Transpyloric approach (by Diderot et al [29], with a timed phylogenetic tree from BEAST used as input for SEIR model simulation. The outputs of the tool are transmission trees (transmission chains) with different branch colours (Figure 3), which denote direct transmission events from person to-person as defined in Transpyloric method [29]. The main difference between the two methods is that Transpyloric uses SIR model (assuming that susceptible individuals move directly to disease after acquiring infection), while the method applied by Eldholm et al [30], applies a SEIR model (assuming that after acquiring infection, susceptible individuals move to exposed or latent state before showing disease symptoms) simulation. The additional of exposed (E) state is important as it indicates a transition or incubation period from infection to disease, which reflect the real world of infectious disease. Limitations and advantages of the Eldhom et al method is similar to those described in Transpyloric method as both depend on a time calibrated phylogeny from other software, such as BEAST as input and use similar assumptions, including within-host diversity and other input parameter values. Compared to other traditional methods, these two use transmission trees with branch colours and vertical arrows to better clarify TB transmission from person-to-person. A major advantage of these methods is that they infer the direction of transmission from the data.
Stimson et al [31], developed a new method, to identify whether the genomes of two MTB isolates are part of a cluster. The method is based on a probabilistic approach that uses the molecular clock rate (SNPs per genome per year), transmission rate (transmissions per year) and transmission cut-off (defined as the cut-off level for the transmission method) to define clusters. Clock rate is the substitution rate, defined as the rate of accumulation of changes in a lineage which depends on both the mutation rate and effects of natural selection [31]. In the development of the method, various assumptions were made. For example, the authors assumed that the population from which samples are drawn is homogeneous unless it is otherwise stated, and that transmission is equally likely between hosts irrespective of factors such as HIV and other co-morbidities. In their approach, they compared their tribalistic method and the SNP threshold method by forming clusters using a range of SNP thresholds and transmission cut-offs. In the comparison of the two methods, authors suggested that the method developed in their study is at least as good at identifying direct transmissions within an outbreak as the SNP threshold method, and typically performs at least slightly better. An advantage of Stimson et al method compared to other methods is that it is more flexible to handle SNPs with different substitution process, variability in the substitution and transmission processes, and it is capable to handle more epidemiological data, such as spatial [31]. One of the weaknesses of the probabilistic method developed by Stimson et al [31], as compared to Bayesian method by Diderot et al [29], is that it does not consider within-host diversity to capture pathogen heterogeneity.
Marker et al [32], and colleagues assessed TB transmission based on SNP distances by determining the number of isolates per isolate that were in a range of ≤10 SNPs referred to as transmission index. They used 10 SNP threshold to infer the number of recently linked cases that considered within a 10-year time period. In the implementation of their method, authors aimed to link each isolate with a continuous parameter which reflects the number of recently linked cases as transmission networks. The networks reflected with a minimum spanning tree which allows the visualization of super-spreaders. They assumed that an isolate with a high transmission index might well be linked to a patient that infected multiple secondary cases. The main difference from SNP threshold approach is that transmission index uses a number of isolates within a maximum range of 10 SNPs to infer the number of recently linked cases, assuming that an isolate with a high SNP distance might well be linked to a super-spreader. Advantage of the transmission index based method by Marker et al is that it has the potential to indicate transmission hotspots within an outbreak scenario and it is independent from a phylogenetic clade definition, which might be difficult to assign due to the close genetic relationship in TB outbreaks and low bootstrap values for small sub-groups at the tips of a tree. Limitation of the transmission index approach is that it is not well stated which software or packages used for SNP distances identification and how spanning tree for transmission networks were constructed.
It is generally believed that the genetic distance between paired isolates (number of SNPs that differ between two sequences) can be used to assess TB transmission [13,19,31]. The method assumes that a genetic distance between strains that is below a specified number of SNPs suggests recent transmission. Although it has been widely applied to measure recent TB transmission, SNP thresholds vary substantially across studies and it is not well understood which value or range of SNP thresholds should be used to infer TB transmission and how such thresholds might vary across settings [31,42]. While SNP-threshold based methods are technically easy to apply and make intuitive sense, they could suggest transmission incorrectly in high incidence settings with endemic strains but no epidemiological links [53]. For example, one study [7], from a high incidence TB setting found that several TB patients with genetic distances of ≤5 SNPs lacked epidemiological links, indicating casual transmission or missing source cases.
While most studies described here applied the SNP threshold method without including drug resistance conferring mutations, studies [19-26] included drug resistance mutations in as an additional criterion to infer TB transmission. In these studies, it was assumed that strains that emerged from the same monophyletic group would have smaller genetic distance and the same drug resistance conferring mutations. However, mixed infection, antibiotic treatment pressure, and the convergent evolution of common drug resistance mutations complicate the use of drug resistance mutations to infer transmission, particularly in high TB burden settings. Strains that are genetically similar enough to be included in the same cluster may have both common and different drug resistance conferring mutations, implying the presence of both primary and acquired resistance [35,53]. For example, studies indicated the presence of common mutations conferring resistance to first-line drugs amongst all clustered isolates but different mutations resistance to second-line drugs, suggesting the occurrence of both primary and acquisition resistance [8,69].
Newer methods based on Bayesian methods together with epidemiological modelling [29,30], probabilistic approaches [31], or transmission index [32], have been applied to assess TB transmission. While these methods provide a more robust assessment of TB transmission, and in the case of Bayesian methods, take genetic heterogeneity into account, they are more difficult technically to implement. Moreover, the Bayesian and epidemiological methods rely on a BEAST analysis, which is dependent on having a large enough genetic diversity within the sample set. Two studies discussed here applied both SNP threshold and Bayesian methods to assess transmission, both indicating the SNP threshold method performed better. While the newly developed Bayesian [29], and probabilistic methods [31], construct transmission trees and clusters to describe recent TB transmission, some of these methods make specific assumptions that might not be relevant to all settings and moreover, are technically difficult to implement. Compared to other methods, the probabilistic method by Stimson et al is more flexible to handle SNPs with different substitution process, and it is capable to handle more epidemiological data, such as spatial. Of newly developed methods, Transpyloric method has been applied in several studies, such as [27,28], as it incorporates within-host diversity and produces informative transmission trees with stars and vertical arrows, indicating the occurrence of transmission events from person-to-person. The main limitation of the Transpyloric based methods is that they assume that all outbreak cases have been sampled and sequenced and that the outbreak has reached its end [33]. In reality, all outbreak cases cannot be sampled as some cases may not be reported to health care. The transmission index based method by Marker et al [32], aimed to identify the number of recently linked cases as transmission networks and visualisation of super-spreaders in a spanning tree. This method assumed that an isolate with a high transmission index (≤ 10 SNP distances) might well be linked to a patient that infected multiple secondary cases. Advantage of the transmission index-based method is that it has the potential to indicate transmission hotspots within an outbreak. However, software or packages used in this method for SNP distances identification and construction of spanning tree for transmission networks are not clarified for utilization in other studies.
In addition to the specific approach used to assess clustering and transmission, underlying bioinformatics pipelines can impact inference of transmission [61]. For example, almost all studies discussed here used a single MTB H37Rv strain as a reference genome for read mapping prior to variant calling. However, one study [18] suggested that since the mutation rate of MTB is very low and stable, this approach may result in limited resolution because it does not take every detectable difference into account, and suggests using a pangenome approach that may detect more variants. In addition to the choice of reference genome, another study suggested that the choice of variant calling can also influence the number of SNPs detected, leading to conflicting transmission inferences, and concluded that measurements of genetic distance and phylogenetic structure depend on variant calling [61]. Here, moves to standardise the TB WGS pipeline could be useful.
In addition to bioinformatics, all WGS based approaches to inferring transmission could face the common biases, including how to disentangling genetic heterogeneity due to different sampling culturing processes prior to WGS and data analysis pipelines in different environments also remains a challenge [31,42,54]. The relatively low genetic diversity (compared to other microbes) is also problematic for transmission models for MTB [68].
Epidemiological contact tracing, based on patient interviews is an important component of investigating TB transmission, and could be combined with WGS [17], or used to validate genotypic methods of assessing transmission. However, the majority of studies reviewed used WGS to infer the possibility of TB transmission without contact tracing, particularly those conducted in high TB burden settings. While contact tracing might be of limited use in high burden settings to interrupt TB transmission, specially designed studies that apply this technique to develop and verify tools to apply to WGS to infer transmission may be useful.
Competing interests: The author declares no competing interests.